量子化学程序原理与功能
Multiwfn入门

Multiwfn入门tips文/Sobereva 2012-Nov-7Multiwfn是一个功能广泛、高效、易用的量子化学波函数分析程序。
写本文的目的是帮助刚接触Multiwfn的人能够在短时间内了解Multiwfn的基本原理以及如何使用。
但此文并不讲解程序操作过程和原理,因为这些内容已经在诸多帖子、程序手册里有详尽描述和示例。
本文着重谈一下应该优先看哪些资料,如何使用手册等问题,使读者明白Multiwfn的使用根本没有什么门槛。
与此同时也提及一些量化刚入门的用户可能会忽略的要点或困惑的问题。
本文内容对应的是Multiwfn 2.6版。
1 对使用者的要求对于量化初学者,Multiwfn当成一个工具作为黑箱来用也可以,但是我还是建议使用者具备一些最基本的理论知识,这样才能避免犯低级错误,才能更透彻地理解程序原理和输出信息的物理意义。
使用者只要仔细读过Levine的Quantum chem istry第五版或第六版(或具有相同级别的知识),就已经足够了,结合手册中对各个功能的理论的讲解,就完全能够理解Multiwfn涉及的全部功能的原理了。
绝大部分Multiwfn的功能运算效率都很高,而且支持并行,在普通个人双核机子上运行就已经挺快了。
完全没必要弄到服务器上去执行。
2 程序的下载、安装、执行和引用Multiwfn最新版本的可执行文件、源代码和手册pdf文档在 的首页上点击相应链接即可下载,老版本可以点击download标签然后下载。
文件名上带bin 或binary的表明是已编译好的可执行文件,src代表源代码文件包。
在首页的Recent update history栏目中可以看到最新版本更新了哪些内容。
在每个正式版本发布之前,有可能也把正在开发的临时版本挂在这个栏目上。
临时版本未经全面测试,手册也没写全,但是已经实现了更新历史上提到的最新功能和改进。
如果想尝鲜可以试试。
Multiwfn更新比较快。
新版本中总会不断添加新的有用的功能、改善界面设计使之更好用、修复各种bug、提升运行效率。
理论化学中的量子化学计算

理论化学中的量子化学计算随着科技的发展和人们对物质本质认识的深入,化学的理论研究也越来越受到人们的关注。
量子化学计算作为化学理论研究的一种方法,已经成为当前化学界研究的热点之一。
本文将介绍量子化学计算的基本概念、方法和应用,并从中探讨其在化学研究中的作用和价值。
量子化学计算是指利用量子力学原理和计算机技术对分子或化学反应进行数值模拟和计算的一种方法。
其关键在于通过计算机技术对分子及其电子结构进行计算,从而研究分子的结构、性质及其与周围环境的相互作用等问题。
以往化学研究主要通过实验手段进行,往往需要大量时间和物质资源,而量子化学计算则可节约研究成本和时间,提高研究效率。
量子化学计算的基本概念1.量子力学:是描述微观世界中的物理规律的一种物理学理论。
量子力学提出了“波粒二象性”和“不确定原理”等基本概念,可以对分子及其电子结构进行描述和计算。
2.分子结构:分子由原子组成,原子之间通过键相连,形成分子的框架结构。
分子的组成和结构在很大程度上决定了分子的性质和用途。
因此研究分子的结构为化学研究提供了基础信息。
3.电子结构:电子是分子中最重要的组成部分,其动态行为直接决定了分子的性质和反应。
因此研究分子的电子结构及其变化规律是一项重要研究内容。
量子化学计算的基本方法1.量子化学基本模型:量子化学计算主要基于分子轨道理论,即分子内电子排布情况决定了分子的性质和反应。
常用的计算模型有:HF计算、密度泛函理论(DFT)计算、MP2计算等。
2.分子坐标和电荷分布:分子坐标包括各原子的空间位置和连接方式,而电荷分布包括电子的整体分布以及电子对于不同原子之间的分配。
利用计算机程序对分子进行描述和计算,需要对分子间的坐标和电荷分布进行精确的描述和设置。
3.计算方法:现有的计算方法分为两类:一种是量子力学方法,包括密度泛函理论、哈特里-福克(Hartree-Fock)方法、MP2、CC、CASSCF等;另一种是经典力学方法,包括分子力场方法、分子动力学方法、蒙特卡洛方法等。
计算化学实验_分子结构模型的构建及优化计算
计算化学实验_分⼦结构模型的构建及优化计算实验9 分⼦结构模型的构建及优化计算⼀、⽬的要求1.掌握Gaussian 和GaussView程序的使⽤。
2.掌握构建分⼦模型的⽅法,为⽬标分⼦设定计算坐标。
3.能够正确解读计算结果,采集有⽤的结果数据。
⼆、实验原理量⼦化学是运⽤量⼦⼒学原理研究原⼦、分⼦和晶体的电⼦结构、化学键理论、分⼦间作⽤⼒、化学反应理论、各种光谱、波谱和电⼦能谱的理论,以及⽆机、有机化合物、⽣物⼤分⼦和各种功能材料的结构和性能关系的科学。
Gaussian程序是⽬前最普及的量⼦化学计算程序,它可以计算得到分⼦和化学反应的许多性质,如分⼦的结构和能量、电荷密度分布、热⼒学性质、光谱性质、过渡态的能量和结构等等。
GaussView是⼀个专门设计的与Gaussian配套使⽤的软件,其主要⽤途有两个:构建Gaussian的输⼊⽂件;以图的形式显⽰Gaussian计算的结果。
本实验主要是借助于GaussView程序构建Gaussian的输⼊⽂件,利⽤Gaussian程序对分⼦的稳定结构和性质进⾏计算和分析。
三、软件与仪器1.软件:Gaussian03、GaussView计算软件,UltraEdit编辑软件。
2.仪器:计算机1台。
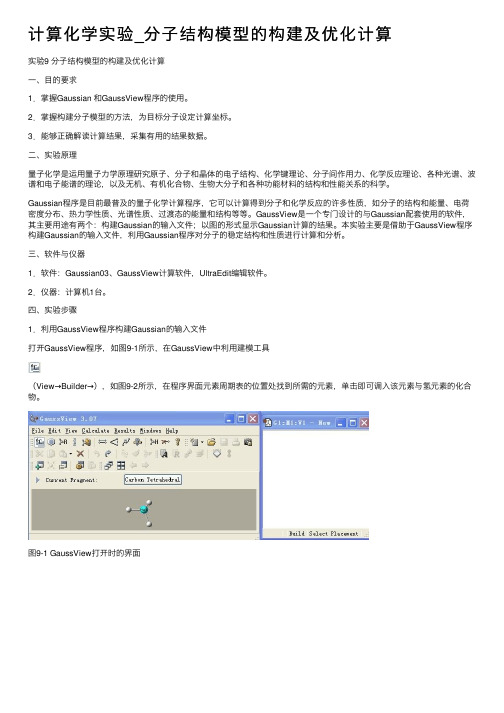
四、实验步骤1.利⽤GaussView程序构建Gaussian的输⼊⽂件打开GaussView程序,如图9-1所⽰,在GaussView中利⽤建模⼯具(View→Builder→),如图9-2所⽰,在程序界⾯元素周期表的位置处找到所需的元素,单击即可调⼊该元素与氢元素的化合物。
图9-1 GaussView打开时的界⾯图9-2点击Builder及双击图标后出现的元素周期表窗⼝图若要构建像⼄烷这样的链状分⼦,需要先点击⼯具栏中的按钮,常见的链状分⼦就显⽰在新打开的窗⼝中,如图9-3所⽰。
图9-3 常见链状官能团窗⼝图若要构建像苯、萘等环状结构的分⼦结构,需要双击⼯具栏中的按钮,常见的环状有机分⼦就显⽰在新打开的窗⼝中,如图9-4所⽰。
量子化学概述

量子化学概述
标题:量子化学概述
正文:
量子化学是一门研究原子和分子的物理性质及其变化的学科。
它基于量子力学的原理,通过数学计算和理论模型来描述和解释原子与分子的结构、性质和反应。
首先,量子化学研究的核心是原子和分子的波函数。
根据薛定谔方程,波函数可以描述系统的状态和性质。
通过求解薛定谔方程,我们可以得到原子和分子的能量、振动频率、轨道结构等信息。
其次,量子化学研究的一个重要应用领域是分子结构预测。
通过计算,可以确定分子的几何构型、键长和键角等。
这对于理解分子的稳定性和反应活性具有重要意义。
同时,量子化学还可以预测分子的光谱性质,如吸收光谱和拉曼光谱,进一步揭示物质的结构特征。
此外,量子化学也在解释化学反应机制、催化过程和原子与分子间的相互作用方面发挥重要作用。
通过量子化学计算,可以研究反应的速率常数、中间体的形成和反应物的能垒等。
这对于设计新的催化剂和理解化学反应动力学有着重要意义。
最后,量子化学还与实验研究相结合,通过理论模拟和实验验证相互印证,共同推动科学的进展。
量子化学的发展也为材料科学、生物化学和药物设计等领域提供了理论基础和研究方法。
总结起来,量子化学是一门基于量子力学原理的学科,通过数学模型和计算方法研究原子和分子的性质和反应。
它在预测分子结构、解释化学反应机制和催化过程等方面发挥着重要作用,为化学和相关领域的研究提供了关键的理论支持。
量子化学是以量子力学为理论基础

量子化学是以量子力学为理论基础量子化学综述一量子化学简介量子化学是以量子力学为理论基础,以计算机为主要计算工具来研究物质的微观结构与宏观性质的关系科学,用以解释物质和化学反应所具有的特性的内在本质及其规律性。
1926年,薛定谔成功地解决了量子态ψ( r , t)是如何随时间演化及各种情况下求出波函数的问题,提出了著名的薛定谔方程。
1927年,化学家Heitler和London等人成功地利用量子力学理论解释了H分子的形2成,开辟了用量子力学方法研究分子中电子行为的广阔领域,标志着量子化学的开始。
随着量子化学基础理论和计算方法的发展,不但使其成为解释化学现象微观本质的强有力工具,而且,使通过量子化学计算来预测化合物性能成为可能。
量子化学是理论化学的一个分支学科,是将量子力学的原理和方法应用到化学问题研究中而产生的一门学科,经过化学家们的努力,量子化学理论和计算方法在近几十年来取得了很大的发展,在定性和定量地阐明许多分子、原子和电子尺度级问题上已经受到足够的重视。
目前,量子化学已被广泛应用于化学的各个分支以及生物、医药、材料、环境、能源、军事等领域,取得了丰富的理论成果,并对实际工作起到了很好的指导作用。
量子化学可分基础研究和应用研究两大类,基础研究主要是寻求量子化学中的自身规律,应用研究是利用量子化学方法处理化学问题,用量子化学的结果解释化学现象。
二现代价键理论计算方法1 价键波函数的计算在价键方法中一个重要的性质是使用了非正交轨道,所有的N!项都对矩阵元有贡献,至今为止仍然没有高效的算法来计算Hamiltonian和重叠矩阵元,这就是价键理论中著名的"N!"困难。
计算Hamiltonian矩阵元和重叠矩阵元有两种方法:一种是经典的行列式展开方法,另一种则是对不变式方法。
a .经典的行列式展开一个HLSP波函数可以表达2n个Slater行列式的线性组合:n为共价键的个数,D(Ωk)为D(Ωk)的Slater行列式,P i是第i个共价键中交换该键的两个成键电子的算符。
量子化学基本原理与应用

量子化学基本原理与应用
量子化学是一种基于量子力学原理的化学研究方法,它可以用来解释和预测分子的结构、反应和性质。
量子化学的基本原理是基于薛定谔方程,它描述了分子中电子的运动和相互作用。
通过求解薛定谔方程,可以得到分子的波函数,从而计算出分子的能量、电荷分布和反应动力学等信息。
量子化学的应用非常广泛,它可以用来研究分子的结构、反应机理、光谱学和材料科学等领域。
例如,在药物设计中,量子化学可以用来预测分子的活性和选择性,从而指导药物的设计和优化。
在材料科学中,量子化学可以用来研究材料的电子结构和光学性质,从而指导材料的设计和合成。
量子化学的计算方法包括从头算和半经验方法。
从头算是指从基本的物理原理出发,通过数值计算求解薛定谔方程,得到分子的波函数和能量等信息。
半经验方法则是基于实验数据和经验参数,通过简化模型来计算分子的性质。
从头算方法的精度更高,但计算量也更大,适用于小分子和简单体系的研究。
半经验方法则更适用于大分子和复杂体系的研究。
量子化学是一种非常重要的化学研究方法,它可以用来解释和预测分子的结构、反应和性质。
随着计算机技术的不断发展,量子化学的应用也越来越广泛,为化学研究和应用带来了巨大的推动力。
量子化学-3.1

Value
0.001235
Threshold
0.000450
Converged?
NO
RMS
Force
0.000234
0.103483
0.000300
0.001800
YES
NO
Maximum Displacement
RMS
Displacement
0.012763
0.001200
NO
Maximum Force:力的收敛标准是0.00045;
• 势能面把能量与分子的每个何结构联系起来 • 这对应于在解分子体系的Schrö dinger 方程时采用了
Born-Oppenheimer近似
• 因此, 势能面是Born-Oppenheimer(核固定)近似的必然结果
对于体系的最小点或鞍点,其能量的一阶导(也就是梯度) 为零。所有成功的优化都会找到一个极小点。
RMS Force:力的均方根的收敛标准为0.0003;
Maximum Displacement:位移的收敛标准为0.0018; RMS Displacement:位移均方根的收敛标准是0.0012。
例:乙烷的优化(计算执行路径行:# B3LYP/6-31G* Opt)
输出结果的解释:
2.3.2 频率计算(Freq)
2.3.3 单点算(SP Calculation)
单点能计算是指在给定的构型上计算分子的能量和相关性质 (包括电荷密度、偶极距和分子轨道等)。和频率计算不同 的是,单点能计算可以在由较低级别计算得到的优化构型上 进行更高级别的能量计算。
C (6-31G**)
I (Lanl2DZ)
PhI (6-311++G**-lanl2dz)
量子化学程序Gaussian原理与功能

化学的对象及其理论描述
分子
原子核 (经典力学)
原 子
芯
原子
内层电子
核外电子 (量子力学)
价层电子
分子的Schrödinger方程
整体 Hy(核,电子)=E•y(核,电子)
Born-Oppenheimer近似 质子的质量是电子的1836倍
只剩下电子 Hy电子=E•y电子
HF与DFT的能级比较: H2O, 6-31G(d,p)
4. 基组的近似与选取
MO-LCAO使得HF方程变成代数方程
恰当地说,它是一种数学技巧与化学认 识的综合。
在数学方面,用自洽场方法(类似于最小 二乘法)去拟合出分子轨道各个原子轨道 的系数(类似于直线的斜率和截距)。
原则上,所用的函数可以是任一组完备 函数集合。但这种完备集合是无穷大的。
交换相关泛函
局域密度近似(LDA):EXC[r(r)] 梯度校正(GGA): EXC[r (r), r (r) ] 梯度校正并未增加很多计算量,因此一般都
使用到梯度校正
常用泛函有:B3LYP(杂化),BLYP,PBE
所有泛函都包含几个经验参数,由小分子拟 合得到。
从计算结果与实验结果的对比上确定对体系 合适的交换相关泛函, 如几何结构
量子化学程序Gaussian原理与功能
量子化学程序Gaussian的原理和功能
I. 模型化学概况 II. 波函数的确定 III. Windows版G03W与GaussView的使用 IV. 模型化学
模型化学
量子化学方法 <500
准确, 化学键
分子力学方法 ~1万—100万 粗略, 只有结构二者杂源自方法小数值=大数值-大数值