量子化学计算方法试验
实验37量子化学计算

实验37 量子化学计算一、目的要求1.通过计算机操作,了解如何运行量子化学应用程序及编制数据输入文件。
2.用HMO 法计算共轭分子的电荷密度、键级及自由价,作出分子图,井预测分子的化学性质。
3.用HMO 法研究若干种富勒烯分子,比较不同异构体之间稳定性。
3.用半经验方法计算富勒烯分子总能量、生成热、前线轨道能量。
二、原理计算化学是化学、物理学和计算机科学等学科的交叉学科。
计算化学包括各种数学计算方法,主要有两大范畴:分子力学和量子化学。
分子力学应用传统的牛顿力学和统计热力学方法主要研究分子的运动规律(如平动、转动和振动等)。
量子化学根据分子轨道理论,通过求解分子体系的薛定谔方程,得到分子轨道波函数和相应的能量以及分子的电子结构和体系总能量,这是量子化学计算的基本内容。
并通过进一步计算得到电子的电离能、电荷密度分布、偶极矩、键级、几何构型以及分子的势能面等等信息。
量子化学计算方法根据所用的近似波函数形式不同可分为两大类:一类是用slater 行列式表示的体系近似波函数,用自治场方法(SCF)求解Hartree-Fock-Roothann 方程。
从头计算法(ab initio)是这类方法中最为严格的一种,在非相对论近似、Born-oppenheimer 近似和轨道近似基础上,对所有的积分既不忽赂,又不用经验参数,严格求解方程,但计算工作量很大,只适用于较小的分子。
半经验方法计算方法包括推广Huckel 、MND0、MNDO-d 、MINDO/3、AM1、PM3等多种方法。
最早是Pople 在20世纪60年代提出的,将从头计算法中所有包含不同原子轨道的积分(称微分重叠)都予以忽略,某些积分用经验参量代替,基态原子轨道简化为只包含价轨道,内层轨道看作原子实,可应用于较大分子性质的计算。
另一类体系的近似被函数用单电子函数(分子轨道)的简单乘积表示,HMO 法就是这一类简单分子轨道法,适用于计算共扼分子,虽然近似程度大,结果不太精确,但对定性讨论有机共轭分子同系物性质规律很有用,多年来一直是理论有机化学经常使用的半经验计算方法。
量子化学计算方法试验

量子化学计算方法试验1. 应用量子化学计算方法进行计算的意义化学是一门基础学科,具有坚实的理论基础,化学已经发展为实验和理论并重的科学。
理论化学和实验化学的主要区别在于,实验化学要求把各种具体的化学物质放在一起做试验,看会产生什么新的物质,而理论化学则是通过物理学的规律来预测、计算它可能产生的结果,这种计算和预测主要借助计算机的模拟。
也就是说,理论化学可以更深刻地揭示实验结果的本质并阐述规律,还可以对物质的结构和性能预测从而促进科学的发展。
特别是近几年来,随着分子电子结构、动力学理论研究的不断深入以及计算机的飞速发展,理论与计算化学已经发展成为化学、生物化学及相关领域中不可缺少的重要方向。
目前,已有多种成熟的计算化学程序和商业软件可以方便地用于定量研究分子的各种物理化学性质,是对化学实验的重要的补充,不仅如此,理论计算与模拟还是药物、功能材料研发环境科学的领域的重要实用工具。
理论化学运用非实验的推算来解释或预测化合物的各种现象。
理论化学主要包括量子化学,(quantum chemistry)是应用量子力学的基本原理和方法研究化学问题的一门基础科学。
研究范围包括稳定和不稳定分子的结构、性能及其结构与性能之间的关系;分子与分子之间的相互作用;分子与分子之间的相互碰撞和相互反应等问题。
量子化学可分基础研究和应用研究两大类,基础研究主要是寻求量子化学中的自身规律,建立量子化学的多体方法和计算方法等,多体方法包括化学键理论、密度矩阵理论和传播子理论,以及多级微扰理论、群论和图论在量子化学中的应用等。
理论与计算化学的巨大进展,正使化学学科经历着革命性的变化。
今天的理论与计算化学几乎渗透到现代一切科技领域,与材料、生物、能源、信息和环保尤为密切,理论化学的应用范围将越来越广。
理论与计算化学逐步发展成为一门实用、高效、富有创造性的基础科学,在化学、生物学等领域的影响越来越显著,且与日剧增。
2. 应用量子化学计算方法进行计算的目的(1)了解量子化学计算的用途。
量子化学计算实验详解
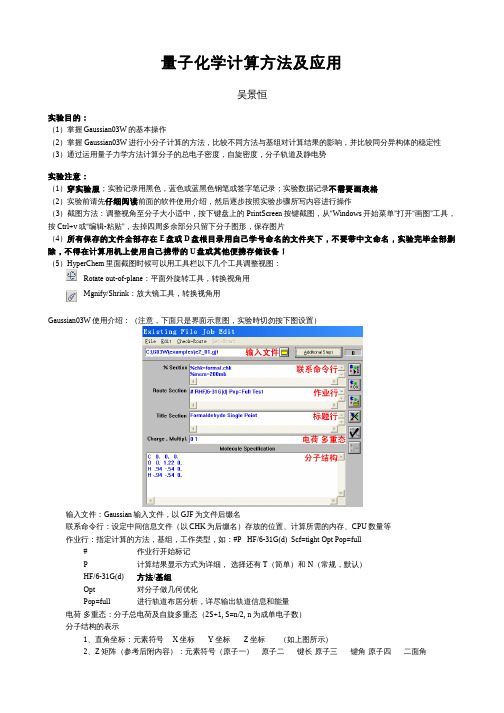
量子化学计算方法及应用吴景恒实验目的:(1)掌握Gaussian03W的基本操作(2)掌握 Gaussian03W进行小分子计算的方法,比较不同方法与基组对计算结果的影响,并比较同分异构体的稳定性(3)通过运用量子力学方法计算分子的总电子密度,自旋密度,分子轨道及静电势实验注意:(1)穿实验服;实验记录用黑色,蓝色或蓝黑色钢笔或签字笔记录;实验数据记录不需要画表格(2)实验前请先仔细阅读前面的软件使用介绍,然后逐步按照实验步骤所写内容进行操作(3)截图方法:调整视角至分子大小适中,按下键盘上的PrintScreen按键截图,从“Windows开始菜单”打开“画图”工具,按Ctrl+v或“编辑-粘贴”,去掉四周多余部分只留下分子图形,保存图片(4)所有保存的文件全部存在E盘或D盘根目录用自己学号命名的文件夹下,不要带中文命名,实验完毕全部删除,不得在计算用机上使用自己携带的U盘或其他便携存储设备!(5)HyperChem里面截图时候可以用工具栏以下几个工具调整视图:Rotate out-of-plane:平面外旋转工具,转换视角用Mgnify/Shrink:放大镜工具,转换视角用Gaussian03W使用介绍:(注意,下面只是界面示意图,实验時切勿按下图设置)输入文件:Gaussian输入文件,以GJF为文件后缀名联系命令行:设定中间信息文件(以CHK为后缀名)存放的位置、计算所需的内存、CPU数量等作业行:指定计算的方法,基组,工作类型,如:#P HF/6-31G(d) Scf=tight Opt Pop=full#作业行开始标记P 计算结果显示方式为详细, 选择还有T(简单)和 N(常规,默认)HF/6-31G(d) 方法/基组Opt对分子做几何优化Pop=full进行轨道布居分析,详尽输出轨道信息和能量电荷 多重态:分子总电荷及自旋多重态(2S+1, S=n/2, n为成单电子数)分子结构的表示1、直角坐标:元素符号X坐标Y坐标Z坐标(如上图所示)2、Z矩阵(参考后附内容):元素符号(原子一)原子二键长原子三键角原子四二面角GaussView使用介绍:主窗口(左),分子窗口(中)原子片段窗口(右):实验步骤:一、计算准备打开GaussView,在新建的分子窗口中画出给定的分子,点击右键选择Lables显示原子序号点击File – Save...,把分子保存为mol.gjf文件;用记事本打开mol.gjf文件,根据分子的对称性修改分子的Z矩阵,为相同环境的原子设置相同的键长并给出名称及初始值,如丙二烯的初始Z-矩阵:CC 1 B1H 2 B2 1 A1H 2 B3 1 A2 3 D1C 4 B4 2 A3 1 D2H 5 B5 4 A4 2 D3H 5 B6 4 A5 2 D4B1 1.35520000B2 1.07000000B3 1.07000000B4 3.37362449B5 1.07000000B6 1.07000000A1 120.22694612A2 119.88652694A3 44.15219753A4 118.94699673A5 118.62610381D1 -180.00000000D2 0.00000000D3 -99.10658593D4 98.97664937由于氢3,氢4与碳2的键长和氢6,氢7与碳5的键长均相等,所以B2、B3、B5、B6均可设定为键长CH(自定义名字,注意所有字母都用大写!),把下面的B2改为CH并把B3、B5、B6删除(数值不同不要紧,后面已为其给出相同的键长初始值);另外为了方便辨认把B1统一改为键长CC,B4改为键长CC2,键角二面角可无视;改好的Z矩阵如下:CC 1 CCH 2 CH 1 A1H 2 CH 1 A2 3 D1C 4 CC2 2 A3 1 D2H 5 CH 4 A4 2 D3H 5 CH 4 A5 2 D4CC 1.35520000CH 1.07000000CC2 3.37362449A1 120.22694612A2 119.88652694A3 44.15219753A4 118.94699673A5 118.62610381D1 -180.00000000D2 0.00000000D3 -99.10658593D4 98.97664937注意内坐标所有变量均在后面给出初始值,变量设值不能重复,数值必须带小数点,改好后保存,用GaussView点击File – Open尝试打开mol.gjf文件,如果打开有误请再检查和修改;打开无误后,把分子结构和原子编号,以及修改后的Z矩阵记录下来,并标明各键长二、量子力学方法几何优化计算比较(1)打开Gaussian03W,点击File – Open...打开刚才保存的mol.gjf文件,作如下修改(不需区分大小写):%Section部分全部删除,不需要关键词Route Section部分修改为:#T HF/3-21G Opt=(Z-matrix,Maxcycle=300) NoSymmTitle Section部分可不修改或改为自定义标题,如:AlleneCharge & Multipl.部分不需修改,按默认值:0 1Molecule Specification部分会读取分子Z矩阵,不需修改,注意开头不要有空行点击File – Save Job保存Gaussian输入文件;点击右边的Run按钮,保存输出文件为mol.out,点击“保存”开始计算作业;当计算窗口Run Progress上显示“Processing Complete.”且输出窗口末尾出现”Normal termination”字样时,表示计算正常结束(如计算出现“Error termination”或中途结束点击确定,点击File – Modify...正确修改输入文件后继续)计算结束后点击View – Editor -> Output File查看输出文件,从末尾开始找到”Job cpu time”记录计算时间;在倒数约第十行附近找到并记录能量值HF=...;往上找到”Dipole moment”记录总偶极矩Tot=...(不需要记录xyz方向上的偶极矩);找到”Optimized Parameters”一栏下”Value”一列,记录优化后的各键长值(只需要键长)(2)点击File – Modify...在Route Section一栏把3-21G 基组改为6-31G*,重复上述计算并记录各上述数据(3)点击File – Modify...基组保持修改后的6-31G*不变,把方法从HF改为MP2重复上述计算并记录各数据(能量数据为MP2=...),记录后把保存的mol.out复制出来备用(该步骤完成后在后面的计算进行时可顺便进行下面第四大点的步骤操作以节省时间)(4)点击File – Modify...,基组保持6-31G*不变,把MP2方法改为B3LYP,计算并记录上述数据(能量值依然为HF=...),并找到”Mulliken atomic charges”记录各原子电荷(原子环境相同的只需记下一个值)(5)点击File–Modify...在Route Section最后加上”Freq”关键词,即修改为“#T B3LYP/3-21G Opt=(Z-matrix,Maxcycle=300) NoSymm Freq”进行计算,通过查找并记录1Atm压力下的零点能(Zero-point correction)、0K内能U0K(Sum of electronic and zero-point Energies)、常温下内能U298.15K(Sum of electronic and thermal Energies),焓H298.15 K(Sum of electronic and thermal Enthalpies) 以及吉布斯自由能G298.15 K(Sum of electronic and thermal Free Energies),通过实验教材上的公式求出熵值并换算成kJ/mol写在数据记录上三、同分异构体稳定性比较用GaussView画出二氯乙烯的两种不同构型的同分异构体(二氯乙烯共有三种异构体,只需其中任意两个即可),分别保存为GJF文件后用记事本打开,复制Z-矩阵(不需要作出改动);在Gaussian03W点击File – Modify...把复制的Z 矩阵贴到Molecule Specification部分,修改Route Section部分为“#T B3LYP/6-31G* Opt(Maxcycle=300)”进行几何优化计算,记录最后所得能量值(只需要HF=...的能量值)四、描绘分子性质(1)打开GaussView软件,点击File – Open...在下拉菜单中选择Gaussian Output Files (*.out *.log)找到先前在第二大步第(3)小步保存的mol.out文件打开,然后点击Save...在下拉菜单中选择MDL Mol Files (*.mol)保存为mol.mol文件打开Hyperchem,点击File – Open...在下拉菜单中选择MDL Mol (*.mol),打开刚才保存的mol.mol文件点击Setup – Semi-empirical,选择“CNDO”作为计算方法;点击Options...,如图进行设置:点击OK,然后点击Compute - Single Point进行单点能计算,从状态栏(窗口最下面)读出并记录能量及梯度数值;下面作图中如Plot Molecular Properties Graph或Orbitals 为灰色,如上再进行一次单点能计算点击Display – Labels...在Atoms一栏选择Charge后点击OK,同上记录各不同类型的原子电荷(2)点击Compute - Plot Molecular Properties Graph,选择Electrostatic Potential及3D Isosurface,在Isosurface Rendering标签Rendering一栏中选择Translucent surface,点击OK绘制出三维静电势图,截图保存为3D_EP.png(截图及视角转换参照实验注意第五点,未选中工具栏视角转换工具前切勿在窗口点击任何按键!)(3)点击Compute - Plot Molecular Properties Graph,选择Total Charge Density,选择2D Contours绘制三维总电荷密度图,保存为2D_TD.png,在下方方框中输入0”,选择3D-Isosurface,点击OK绘制HOMO分(4)点击Compute – Orbitals,选中“HOMO-”“子轨道图,截图存为HOMO.png(5)点击Setup – Semi-empirical – Options...更改Total charge为1,Spin multiplicity为2,确定后点击Compute - Single Point再进行一次Single Point单点能计算,记录能量和梯度值;点击Compute - Plot Molecular Properties Graph,选择“Spin density”显示三维的单电子自旋密度图,存为3D_SD.png实验结束,检查下面数据是否已被正确记录:1、分子结构及修改后的Z矩阵表示2、分别记录 HF/3-21G, HF/6-31G*, MP2/6-31G*, B3LYP/6-31G*计算水平下分子几何优化后各项性质(计算时间、能量、总偶极矩以及各优化键长)3、B3LYP/6-31G*计算水平下分子的零点能、零度内能、内能、焓、吉布斯自由能和熵值4、MP2/6-31G*方法及CNDO方法所得不同环境的原子电荷5、二氯乙烯分子两个同分异构体在B3LYP/6-31G*水平下计算的能量数值6、分子各性质图像打开”网上邻居-综合 在 Zh00 上-2012-物化计算机实验”,找到以当天日期命名的文件夹,在下面新建以自己学号命名的文件夹,把3D_EP.png, 2D_TD.png, HOMO.png, 3D_SD.png复制到里面,把原始数据记录取至前台检查签名(原始数据记录请务必写上姓名!),签名后在前台用U盘把自己的实验图片复制下来或发送到自己邮箱里面(教师用计算机学生不得操作!)实验完毕删除自己在用机上所有留下的有关文件,关闭计算机,收拾桌椅并带好个人携带物品离开实验室实验报告:一、实验原始数据记录应附在实验报告的最后,不能直接作为实验报告的内容部分二、实验图片应打印好作为实验报告的内容部分并标上图片标题和注释,不能附在实验报告的最后三、实验报告所有数据必须用表格形式列出,并应对所有已记录的数据进行分析,所有能量在实验报告中均换算成单位kJ/mol(1Hartree=2625.5 kJ/mol),此外还应包括以下内容:1、查阅文献,查找所做分子的对应键长实验测量数值或使用更准确的理论计算方法所得数据2、比较并分析相同基组下HF,MP2,B3LYP方法的准确程度以及所耗时间3、比较并分析不同基组(3-21G, 6-31G*)下HF方法的准确程度4、比较并分析使用不同方法及基组所得能量数值的大小关系5、比较各方法所得的偶极矩及原子电荷,分析使用不同方法和基组下的计算结果及其合理性6、分析二氯乙烯同分异构体的稳定性大小四、思考题(连同给定的书本上的思考题写入实验报告):1、使用直角坐标和Z矩阵各有哪些优劣?考虑对称性会为计算带来哪些好处和坏处?2、除了给定电荷以外,为什么还要给定分子的自旋多重态?它能反映体系的哪些性质?3、你觉得该实验比较同分异构体的稳定性大小的方法结果可靠吗?请说明理由4、如果有人认为该实验比较同分异构体的稳定性大小的方法不够准确,你会有什么合理的方法进行改进?5、量子力学方法是怎样得到各原子的电荷的?各有哪些方法计算电荷?不同方法所得的电荷数值是否具有可比性,能否用于进行定性,半定性或定量分析?6、静电势、电荷密度、自旋密度和分子轨道都能体现分子内电子分布请况,请说出它们的具体定义,联系及区别7、本次计算在Gaussian03W中用到的各项关键词都是什么意思?试解释在该次实验中为什么使用到这些关键词附:Z矩阵采用采用内坐标(键长,键角和二面角)输入,便于保持分子的对称性,如NH3(对称性为C3V):NH1HNH1HN2 HNHH1HN3 HNH2DHNHN 1.HNH 109.47DNH-120.根据对称性设值可以减少计算量,上述内坐标根据对称性定义了三个变量分别为N-H键键长,H-N-H键角及四个原子所成的二面角,并在后面给出了它们的初始值。
量子化学计算的方法和应用研究

量子化学计算的方法和应用研究量子化学计算是一种利用量子力学原理模拟和计算化学性质的方法。
它已经成为现代化学研究中的重要工具,广泛应用于药物设计、催化剂开发、新材料设计等领域。
本文将介绍量子化学计算的基本原理、常用方法以及其在不同领域的应用研究。
量子化学计算的基本原理是基于量子力学的波函数。
波函数描述了系统的量子态,通过求解薛定谔方程可以得到波函数的信息,从而推导出分子的能量、电子结构以及反应动力学等信息。
因为薛定谔方程的求解是非常困难的,所以量子化学计算中使用了一系列的近似方法。
其中,最常用的方法之一是密度泛函理论(DFT)。
DFT是基于电子密度的理论,通过求解系统的电子密度来近似求解能量和其他性质。
相较于传统的薛定谔方程求解方法,DFT具有较低的计算成本和较好的精度。
因此,它被广泛应用于计算化学的各个领域。
除了密度泛函理论,还有诸如哈特里-福克方法、耦合簇理论等在量子化学计算中具有重要地位的方法。
这些方法在处理不同类型的分子和化学反应时,都有其特点和优势。
根据研究的需要,选择合适的方法进行计算可以更好地揭示分子的性质和反应机制。
在药物设计方面,量子化学计算可以用来研究分子的构象空间、理解药物与受体之间的相互作用、优化药物分子的性质等。
通过计算,可以预测分子的活性、选择性和毒性等特性,从而为药物的设计和优化提供指导。
此外,量子化学计算还可以揭示药物化学反应的机理和动力学,为药物合成工艺的优化提供理论支持。
催化剂是化学反应中常用的协同剂。
通过量子化学计算,可以研究催化剂表面的活性位点、反应机理以及吸附动力学等。
这些信息对于理解反应机制、优化催化剂设计以及预测反应活性具有重要意义。
基于量子化学计算的研究可以辅助实验设计新型催化剂,并提供对其活性、稳定性和选择性的理论解释。
新材料的发现和设计是实现科学技术进步的重要环节。
量子化学计算在材料科学中的应用涵盖了从材料性质预测到材料设计的各个方面。
通过计算,可以预测材料的电子、光学、力学等性质,从而指导实验设计新型材料。
量子化学计算实验报告

一、实验目的量子化学计算是研究化学键和分子结构的理论方法,通过计算机模拟计算分子的能量、结构、性质等。
本次实验旨在让学生了解量子化学计算的基本原理,掌握常用的计算方法,并通过实验加深对量子化学计算在实际问题中的应用。
二、实验原理量子化学计算基于量子力学的基本原理,通过求解薛定谔方程,得到分子的电子结构,进而分析分子的性质。
常用的量子化学计算方法有:分子轨道理论、密度泛函理论、从头算方法等。
三、实验仪器与材料1. 仪器:计算机、量子化学计算软件(如Gaussian、MOPAC等)2. 材料:实验所需的分子结构文件、计算参数文件等四、实验步骤1. 准备分子结构文件:根据实验要求,选择合适的分子结构,并使用分子编辑软件(如ChemDraw、AVG/AVG Plus等)绘制分子结构图。
2. 设置计算参数:根据实验目的和分子结构,选择合适的计算方法、基组、计算级别等参数。
3. 运行量子化学计算软件:将分子结构文件和计算参数文件导入量子化学计算软件,开始计算。
4. 分析计算结果:查看计算结果,分析分子的能量、结构、性质等。
五、实验结果与分析1. 氧分子(O2)的计算以氧分子(O2)为例,使用Gaussian软件进行分子轨道理论计算。
计算结果如下:- 能量:-14.5466 eV- 结构:O2分子的两个氧原子通过共价键连接,键长为1.207 Å,分子轨道能级顺序为σ2s、σ2s、π2p、σ2p。
2. 苯(C6H6)的计算以苯(C6H6)为例,使用Gaussian软件进行休克尔理论计算。
计算结果如下:- 能量:-6.6284 eV- 结构:苯分子中的六个碳原子通过共轭π键连接,分子轨道能级顺序为π1、π2、π3、π4、π5、π6。
3. 苯乙烯(C6H5CH=CH2)的计算以苯乙烯(C6H5CH=CH2)为例,使用Gaussian软件进行从头算方法计算。
计算结果如下:- 能量:-6.8312 eV- 结构:苯乙烯分子中的苯环与乙烯基通过共价键连接,键长分别为1.396 Å和1.341 Å,分子轨道能级顺序为σ2s、σ2s、π2p、σ2p、π3p、π4p、π5p。
实验37 量子化学计算 操作细节

实验37 量子化学计算1.HMO 法计算平面共轭分子(1) 计算前预先将共轭原子用数字编号。
如苯分子,可按图37-3(a )对所有骨架碳原子进行编号(氢原子不必编号)(2)打开计算机进入WINDOWS 操作系统,在微机上找到HMO 目录(例如:D:\HMO ),用鼠标选择“开始-所有程序-附件-命令提示符”,进入DOS 操作系统界面,在该界面中先输入“CD \”,退到C 盘根目录下,然后输入“D :”进入D 盘,再输入“CD \HMO”,进入HMO 目录。
输入“QBASIC”,进入用QBASIC 语言编辑界面(注:以上每次输入完需按回车Enter 键作为结束)。
按键盘左上角的“Esc”键,退出提示界面。
用鼠标选择“File -Open”,打开“Open”对话框,选择“HMO.BAS”文件,再选择“<OK>”,进入HMO 程序界面。
(3)修改共轭分子中的原子连接信息。
HMO 程序由BASIC 语言编写,其中:在语句10中输入共轭原子连接信息,即输入原子编号不按顺序连接的两个原子的编号。
如图37-3(a)苯分子中的1,6两原子。
在语句15中输入不相连的信息,即原子编号连续,而在分子中不相连的两个原子的编号。
如图37-3(b)中的4,5两原子。
上述数据语句中均以两个0表示输入结束。
如苯分子,按以下形式修改即可:10 DATA 1,6,0,015 DATA 0,0在计算丁二烯分子时,若将四个共轭原子从左至右用1,2,3,4顺序编号,那未语句10和15中均改为0,0即可。
134562(a) 23456(b) 图37-3 苯分子编号示意图(4)数据核对无误后,可按F5功能键运行程序。
由屏幕显示的信息,通过人机对话相继输入参数。
如计算苯分子时,“INPUT THIS CALCULATION INFORMATION?”用大写字母输入分子式“C6H6”;“HOW MANY ATOMS?”输入共轭原子数“6”即可(因为该计算方法只考虑骨架碳原子);“WOULD CHANGE INTEG.<Y/N>?”,询问是否修改积分参数,输入大写字母“N”。
量子化学计算实验详解2014
量子化学计算方法及应用实验目的:(1)掌握Gaussian03W的基本操作,通过计算小分子比较不同方法与基组对结果的影响,并比较同分异构体的稳定性;(2)通过运用量子力学方法计算分子的总电子密度,自旋密度,分子轨道及静电势。
实验注意:(1)穿实验服;实验记录用黑色,蓝色或蓝黑色钢笔或签字笔记录,不需要画表格;(2)实验前请先仔细阅读后附的软件使用介绍,然后逐步按照实验步骤所写内容进行操作;(3)所有保存的文件全部保存在E盘或D盘根目录用自己学号命名的文件夹内,文件不要带中文命名,实验完毕全部删除,不得在计算用机上使用自己携带的U盘或其他便携存储设备!实验步骤:一、计算准备打开GaussView,在新建的分子窗口中画出给定的分子结构,点击右键选择Lables显示原子序号;点击File –Save...,把分子保存为mol.gjf文件;用记事本打开mol.gjf文件,根据分子的对称性修改分子的Z矩阵,为相同环境的原子设置相同的键长并给出名称及初始值,以丙二烯的初始Z矩阵为例:CC 1 B1H 2 B2 1 A1H 2 B3 1 A2 3 D1C 4 B4 2 A3 1 D2H 5 B5 4 A4 2 D3H 5 B6 4 A5 2 D4B1 1.35520000B2 1.07000000B3 1.07000000B4 3.37362449B5 1.07000000B6 1.07000000A1 120.22694612(以下省略...)由于氢3,氢4与碳2的键长和氢6,氢7与碳5的键长均相等,所以B2、B3、B5、B6均可设定为键长CH(自定义名称,注意所有字母都用大写!),把下面的B2改为CH并把B3、B5、B6删除(数值不同不要紧,后面已为其给出相同的键长初始值);另外把B1改为键长CC,B4改为键长CC2,键角二面角可无视;修改后Z矩阵如下:CC 1 CCH 2 CH 1 A1H 2 CH 1 A2 3 D1C 4 CC2 2 A3 1 D2H 5 CH 4 A4 2 D3H 5 CH 4 A5 2 D4CC 1.35520000CH 1.07000000CC2 3.37362449A1 120.22694612(以下省略...)注意内坐标所有变量均在后面给出初始值,变量设值不能重复,修改好后保存,用GaussView点击File – Open尝试打开刚才保存的mol.gjf文件,如果打开有误请再检查和修改。
量子化学计算方法的研究和应用
量子化学计算方法的研究和应用随着科技的发展和数值计算能力的不断提升,量子化学计算成为化学界最热门的领域之一。
该领域涵盖了分子结构和反应的理论研究、材料科学、生物医学等众多领域。
本文将简要介绍量子化学计算的基本原理,涉及计算方法,以及其在实际应用中所扮演的角色。
1. 基本原理量子化学计算是一种基于量子力学原理的计算方法,以计算机模拟方式研究化学现象。
其基本原理是根据薛定谔方程和量子力学原理,结合电子结构理论,通过研究电子分布和运动的行为,预测和解释化学反应、材料结构和性质等现象。
在量子化学计算方法中,常用的基础概念包括分子轨道、基组、哈密顿算符等。
2. 计算方法由于分子中存在大量的电子和原子核相互作用,使量子化学计算涉及的计算难度非常大,需要运用一些高效的方法进行计算。
常用的计算方法有:(1) 原子轨道理论:是一种简单的量子力学计算方法,它把一般分子的电子结构问题转化为一些简单的原子问题,再解决这些问题。
(2) 单电子近似:是计算多电子体系中某个电子在某个原子上的行为时,忽略其他电子的影响。
这种近似可以用来计算分子轨道和电子云的分布。
(3) 矩阵对角化方法:是将大量数据转化为一个矩阵的形式,然后通过对这个矩阵的运算来获得需要的结果。
(4) 希尔伯特空间方法:是将给定问题表示成一个希尔伯特空间中的算子形式,然后通过求解这个算子的本征值和本征向量来得到结果。
3. 应用在实际应用中,量子化学计算主要用于预测和解释化学反应、材料的结构和性质、无机和有机分子的电子结构、光谱、动力学以及生物分子的结构和功能等等。
有很多研究表明,量子化学计算在化学领域中有着广泛的应用。
3.1 分析物质结构和性质量子化学计算可以用来预测分子的幾何構型和分子内的原子價鍵的長短、角度大小、键能和键离子体积等性质。
这种方法可以节约大量实验工作和费用,并且能够为合成新分子提供指导。
3.2 预测化学反应量子化学计算方法可以预测和解释化学反应的机理和反应条件,比如反应速率常数、反应路径和反应物与产物之间的关系等。
量子化学计算的基本流程与实践方法
量子化学计算的基本流程与实践方法量子化学计算是一种基于量子力学理论和计算机模拟的方法,用于研究分子和原子的性质和行为。
它可以帮助科学家理解和预测化学反应、材料性质以及生物分子的结构与功能。
量子化学计算在材料科学、药物设计等领域具有广阔的应用前景。
本文将介绍量子化学计算的基本流程和实践方法。
1. 理论基础量子化学计算基于量子力学理论,利用薛定谔方程描述了分子的波函数演化。
通过求解薛定谔方程,可以获得分子的能量、波函数、电子密度等信息。
量子化学计算可以分为两类:基于从头算(ab initio)的计算和基于半经验方法的计算。
前者是完全基于量子力学原理进行计算,而后者则利用一些经验参数和基础假设简化计算。
2. 基本流程量子化学计算的基本流程包括分子几何优化、基组选择、哈特里-福克(HF)计算、密度泛函理论(DFT)计算、分子轨道分析等步骤。
(1)分子几何优化分子几何优化是为了确定分子的最稳定结构,即分子中原子的最佳位置和键长。
分子几何优化可以使用基于梯度的优化算法,如坐标下降法或共轭梯度法。
通过优化分子的几何结构,可以得到分子的电子能量。
(2)基组选择基组是量子力学计算的基础,用于描述单个原子和原子间相互作用。
选择合适的基组对计算结果的准确性至关重要。
常用的基组包括STO-nG、6-31G(d)和cc-pVTZ等。
不同的基组具有不同的精度和计算复杂度,需要根据具体情况进行选择。
(3)HF计算哈特里-福克方法是一种常见的从头算方法,基于单电子近似和双电子积分计算电子能量。
HF方法通过迭代求解薛定谔方程的自洽场得到分子的电子能量。
然而,HF方法只能处理弱相互作用的分子,对含有强电子相关性的体系效果较差。
(4)DFT计算密度泛函理论是一种基于电子密度的方法,可以处理含有强电子相关性的分子。
DFT方法通过最小化系统的总能量来求解分子的电子结构和性质。
常用的DFT方法包括B3LYP、PBE和TPSS等。
DFT方法相对于HF方法计算速度更快,适用于大分子和复杂体系的计算。
量子计算的量子化学方法与应用探究(一)
量子计算是近年来备受关注的前沿领域之一,其潜在应用广泛而多样。
在量子计算中,量子化学是一个重要的研究方向,它研究如何利用量子计算的原理解决化学领域的问题。
本文将探讨量子计算的量子化学方法及其应用。
一、量子计算的背景与概述量子计算是以量子力学原理为基础的一种新兴计算方法。
与传统计算机使用的比特(bit)不同,量子计算机使用的是量子比特(qubit)。
量子比特具有一些特殊的性质,如叠加态和纠缠态,使得量子计算具备强大的计算能力。
二、量子化学方法的发展历程随着量子计算的兴起,越来越多的研究者开始尝试将其引入到化学领域。
最早的量子化学方法可以追溯到20世纪60年代,当时的计算机能力有限,研究者们使用了一些基于近似方法的量子化学方法,如Hartree-Fock方法和密度泛函理论。
随着计算机硬件和算法的不断改进,量子化学领域也出现了一系列新的方法和理论,如多体微扰论、耦合簇方法和量子化学力学方法等。
三、量子化学方法的原理与实现在量子化学方法中,有两个关键问题需要解决:系统的哈密顿量的构建和求解薛定谔方程。
在构建系统的哈密顿量时,需要考虑体系中的电子和核的相互作用,并考虑多体相互作用,这一步通常需要使用一些数学工具和物理模型进行近似处理。
而求解薛定谔方程则需要使用一些数值方法,如变分法和哈密顿矩阵对角化等。
四、量子化学方法的应用领域量子化学方法在化学领域的应用非常广泛。
首先,量子化学方法可以用于预测化学反应的速率和产物分布,帮助理解和解释实验结果。
其次,量子化学方法可以用于设计新型的催化剂和材料,加速新材料的研发进程。
此外,在药物设计和生物化学研究中,量子化学方法也发挥着重要的作用。
五、量子化学方法的挑战与展望尽管量子化学方法在化学领域的应用取得了一些重要的成果,但仍然存在一些挑战和困难。
首先,目前的量子计算机技术仍然处于发展阶段,计算能力相对有限。
其次,在处理大规模体系时,量子化学方法的计算复杂度非常高,需要大量的计算资源。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
量子化学计算方法试验1. 应用量子化学计算方法进行计算的意义化学是一门基础学科,具有坚实的理论基础,化学已经发展为实验和理论并重的科学。
理论化学和实验化学的主要区别在于,实验化学要求把各种具体的化学物质放在一起做试验,看会产生什么新的物质,而理论化学则是通过物理学的规律来预测、计算它可能产生的结果,这种计算和预测主要借助计算机的模拟。
也就是说,理论化学可以更深刻地揭示实验结果的本质并阐述规律,还可以对物质的结构和性能预测从而促进科学的发展。
特别是近几年来,随着分子电子结构、动力学理论研究的不断深入以及计算机的飞速发展,理论与计算化学已经发展成为化学、生物化学及相关领域中不可缺少的重要方向。
目前,已有多种成熟的计算化学程序和商业软件可以方便地用于定量研究分子的各种物理化学性质,是对化学实验的重要的补充,不仅如此,理论计算与模拟还是药物、功能材料研发环境科学的领域的重要实用工具。
理论化学运用非实验的推算来解释或预测化合物的各种现象。
理论化学主要包括量子化学,(quantum chemistry)是应用量子力学的基本原理和方法研究化学问题的一门基础科学。
研究范围包括稳定和不稳定分子的结构、性能及其结构与性能之间的关系;分子与分子之间的相互作用;分子与分子之间的相互碰撞和相互反应等问题。
量子化学可分基础研究和应用研究两大类,基础研究主要是寻求量子化学中的自身规律,建立量子化学的多体方法和计算方法等,多体方法包括化学键理论、密度矩阵理论和传播子理论,以及多级微扰理论、群论和图论在量子化学中的应用等。
理论与计算化学的巨大进展,正使化学学科经历着革命性的变化。
今天的理论与计算化学几乎渗透到现代一切科技领域,与材料、生物、能源、信息和环保尤为密切,理论化学的应用范围将越来越广。
理论与计算化学逐步发展成为一门实用、高效、富有创造性的基础科学,在化学、生物学等领域的影响越来越显著,且与日剧增。
2. 应用量子化学计算方法进行计算的目的(1)了解量子化学计算的用途。
(2)了解量子化学计算的原理、方法和步骤。
(3)通过一两个计算实例进行量子化学计算的上机操作试验。
(4)学会简单的分析和应用计算结果。
3. 量子化学计算试验的原理量子化学是应用量子力学的规律和方法来研究化学问题的一门学科。
将量子理论应用于原子体系还是分子体系是区分量子物理与量子化学的标准之一。
主要分为:①分子轨道法(简称MO法,见分子轨道理论);②价键法(简称VB法,见价键理论);③密度泛函理论。
以下只介绍分子轨道法。
①分子轨道法:分子体系中的电子用单电子波函数满足Pauli不相容原理的直积(如Slater 行列式)来描述,其中每个单电子波函数通常由原子轨道线性组合得到(类似于原子体系中的原子轨道),被称作分子轨道,分子轨道理论是目前应用最为广泛的量子化学理论方法。
o HF方法:它是原子轨道对分子的推广,即在物理模型中,假定分子中的每个电子在所有原子核和电子所产生的平均势场中运动,即每个电子可由一个单电子函数(电子的坐标的函数)来表示它的运动状态,并称这个单电子函数为分子轨道,而整个分子的运动状态则由分子所有的电子的分子轨道组成(乘积的线性组合),这就是分子轨道法名称的由来。
分子轨道法的核心是哈特里-福克-罗特汉方程,简称HFR方程,它是以三个在分子轨道法发展过程中做出卓著贡献的人的姓命名的方程。
1928年D.R.哈特里提出了n个将电子体系中的每一个电子都看成是在由其余的n-1个电子所提供的平均势场中运动的假设。
这样对于体系中的每一个电子都得到了一个单电子方程(表示这个电子运动状态的量子力学方程),称为哈特里方程。
使用自洽场迭代方式求解这个方程(见自洽场分子轨道法),就可得到体系的电子结构和性质。
哈特里方程未考虑由于电子自旋而需要遵守的泡利原理。
1930年,B.A.福克和J.C.斯莱特分别提出了考虑泡利原理的自洽场迭代方程,称为哈特里-福克方程。
它将单电子轨函数(即分子轨道)取为自旋轨函数(即电子的空间函数与自旋函数的乘积)。
泡利原理要求,体系的总电子波函数要满足反对称化要求,即对于体系的任何两个粒子的坐标的交换都使总电子波函数改变正负号,而斯莱特行列式波函数正是满足反对称化要求的波函数。
将哈特里-福克方程用于计算多原子分子,会遇到计算上的困难。
C.C.J.罗特汉提出将分子轨道向组成分子的原子轨道(简称AO)展开,这样的分子轨道称为原子轨道的线性组合(简称LCAO)。
使用LCAO-MO,原来积分微分形式的哈特里-福克方程就变为易于求解的代数方程,称为哈特里-福克-罗特汉方程,简称HFR 方程。
o CI方法:组态相互作用(Configuration Interaction)方法。
用HF自洽场方法计算获得的波函数和各级激发的波函数为基展开体系波函数。
完全的组态相互作用(Full-CI)是指定基组下最精确的方法,但其计算量约以基函数的阶乘规模增加,目前仅限于对小分子作为Benchmark以检测其他方法的可靠性,在实际应用中常采用截断CI方法,如DCI、SDCI等。
由于截断CI不满足体积加合性(size-extensive),虽然有一些经验方法校正(如Davidson correction),仍然限制了其应用。
o MP方法:多体微扰方法,将多电子体系的总哈密顿算符与Fock算符的差作为体系的微扰项,应用Rayleigh-Schroedinger微扰方法计算。
一级微扰等价于HF,二级微扰可以达到甚至超过DCI方法的精度水平,但计算量(N^5)小于DCI(N^6)。
一般不适用于能级接近简并的体系。
任意阶的MP都是size-extensive的。
o多组态自洽场方法:同时对组态和展开系数进行优化,恰当地应用可以克服截断CI 不满足体积加合性的缺点,并比较地准确处理能级近简并的体系。
o半经验计算方法:Extended Huckel method, AM1,PM3等方法。
在计算过程中根据实验数据,将一些波函数积分用经验常数替代,可以上千倍地减少计算量,采用的经验常数不同,半经验算法的应用范围也不同,应用时需要根据研究体系的具体情况进行选择。
②价键法:它的核心是:两个含有单个电子的原子,若它们电子的自旋方向相反,则通过电子的配对,在这两个原子间形成一个共价键。
③密度泛函理论密度泛函理论:当分子体系各原子核空间位置确定后,电子密度在空间中的分布也确定,可以将体系的能量表示为电子密度的泛函,密度泛函分析变分法求出能量最低时的电子密度分布和体系能量。
研究工作者最重要的是要根据自己的研究课题的需要,先构建和确定需要进行理论研究的物质结够模型,再选定能满足计算要求的应用量子化学计算程序和软件,然后进行量子化学计算分析计算结果,获得所关心或感兴趣的能表征物质性质的信息并加以应用。
实验一利用量子化学计算软件验证分子轨道理论和判断分子点群一、主要仪器设备及软件1、仪器:用于计算的计算机。
2、软件A、建模软件(1) Chemoffice是一款广受化学学习、研究者好评的化学学习工具,据说对大学生学习化学帮助很大。
ChembioOffice 是由CambridgeSoft开发的综合性科学应用软件包。
该软件包是为广大从事化学、生物研究领域的科研人员个人使用而设计开发的产品。
同时,这个产品又可以共享解决方案,给研究机构的所有科技工作者带来效益。
利用ChemBioOffice 可进行化学生物结构绘图、分子模型及仿真、将化合物名称直接转为结构图,省去绘图的麻烦;也可以对已知结构的化合物命名,给出正确的化合物名,等。
(2) GaussView 主要功能有创建三维分子模型,计算任务设置全面支持Gaussian 计算,和显示Gaussian计算结果等。
B、计算软件:(1) Gaussian:量子化学领域最著名和应用最广泛的软件之一,由量子化学家约翰波普的实验室开发,可以应用从头计算方法、半经验计算方法等进行分子能量和结构;过渡态能量和结构;化学键及反应能量;分子轨道;偶极矩;多极矩;红外光谱和拉曼光谱,核磁共振,极化率和超极化率,热力学性质,反应路径等分子相关计算。
可以运行在Windows、Linux、Unix操作系统中运行,目前最新版本为Gaussian 09。
但由于Gaussian Inc. (Gaussian的发展者)排斥其他软件发展者的行为,而引来不少批评;其行为包括逐原开发者约翰波普离开Gaussian Inc. (因而成为学术界其中一件为人齿冷的事件);禁止其他开发者(包括约翰波普)使用Gaussian. (引发起Banned By Gaussian 运动和禁止任何使用者发表比较Gaussian与其他量子化学软件效能的报告等。
(2) Materials Studio:是ACCELRYS 公司专门为材料科学领域研究者所涉及的一款可运行在PC上的模拟软件。
他可以帮助你解决当今化学、材料工业中的一系列重要问题。
支持Windows98、NT、Unix以及Linux等多种操作平台的Materials Studio使化学及材料科学的研究者们能更方便的建立三维分子模型,深入的分析有机、无机晶体、无定形材料以及聚合物。
多种先进算法的综合运用使Material Studio成为一个强有力的模拟工具。
无论是性质预测、聚合物建模还是X射线衍射模拟,我们都可以通过一些简单易学的操作来得到切实可靠的数据。
灵活方便的Client-Server结构还是的计算机可以在网络中任何一台装有NT、Linux或Unix操作系统的计算机上进行,从而最大限度的运用了网络资源。
(3) V ASP是使用赝势和平面波基组,进行第一定律分子动力学计算的软件包。
V ASP中的方法基于有限温度下的局域密度近似(用自由能作为变量)以及对每一MD步骤用有效矩阵对角方案和有效Pulay混合求解瞬时电子基态。
这些技术可以避免原始的Car-Parrinello方法存在的一切问题,而后者是基于电子、离子运动方程同时积分的方法。
离子和电子的相互作用超缓Vanderbilt赝势(US-PP)或投影扩充波(PAW)方法描述。
两种技术都可以相当程度地减少过渡金属或第一行元素的每个原子所必需的平面波数量。
力与张量可以用V ASP很容易地计算,用于把原子衰减到其瞬时基态中。
(4) Gamess-US: 由于免费与开放源码,成为除Gaussian以外,最广泛应用的量子化学软件,目前由Iowa State Uinversity的Mark Gorden 教授的研究组主理。
(5) CASTEP: 为一量子力学为基础的周期性固态材料化学计算的套装软件,此程式由英国剑桥大学卡文迪西(Cavendish)实验室的凝态物理理论组所共同研究开发。