利用Hyperchem软件进行分子结构构建及性质计算
HyperChem软件

HyperChem软件中Script的常用语句注解1. 计算方法及参数设定calculation-method item计算方法设定item: MolecularMechanics, SemiEmpirical, AbInitio, DFT molecular-mechanics-method item分子力学方法设定item: mm+, amber, bio+, opls, amber94, charmm22, tndo optim-max-cycles x优化最大迭代次数x设定optim-convergence goal 优化收敛目标goal设定optim-converged返回是否收敛(true/false)optim-algorithm item 优化算法设定item: PolakRibiere, NewtonRaphson, etc.assign-basisset item abinitio计算基组设定item: 3-21G, STO-3G, etc.semi-empirical-method item半经验计算方法设定item: am1, pm3, extendedhuckel, cndo, indo, zindos, mndod, tndo, etc.accelerate-scf-convergence true/false 是否加速SCF收敛scf-convergence goal SCF收敛目标goal设定max-iterations n SCF计算的最大迭代次数n设定2. 计算结果输出omsgs-to-file filename 将信息输出到filename文件append-omsgs-to-file filename 将信息追加到filename文件omsgs-not-to-file 关闭输出文件query-response-has-tag yes/no 提取的信息回显开关query-value HSV提取系统数据结构中的信息并单行输出HSV: current-file-name, coordinates,dipole-moment, dipole-moment-componentstotal-energy, heat-of-formation,scf-binding-energy, scf-core-energy, scf-electronic-energy start-logging HyperChem运行记录开启stop-logging HyperChem运行记录关闭export-property-file filename将分子轨道信息写入filename文件3. 分子结构及其文件操作file-format item定义分子结构文件格式item: hin, mol, zmt, ent, skc, xyz, ml2open-file filename 读入filename分子结构文件write-file filename 将分子结构写入filename文件menu-build-add-hydrogens 给分子加氢menu-build-model-build 给分子加氢并规格化do-optimization 进行优化操作do-single-point 进行单点计算do-molecular-dynamics进行分子动力学操作4. 屏幕显示设置screen-refresh-period n屏幕刷新周期atom-labels item原子标识设定item: none, number, symbol, name, charge, etc.show-hydrogens yes/no 分子中氢显示开关show-multiple-bonds yes/no分子中重键显示开关request string弹出窗口并显示由双引号表达的字符串string 5. 其它read-script filename读取名为filename的script文件exit-script 退出script实例: example.scrcalculation-method molecular-mechanicsmolecular-mechanics-method mm+optim-max-cycles 2048optim-convergence 0.1optim-algorithm PolakRibiereomsgs-to-file results.txtquery-response-has-tag nofile-format molopen-file 000A-0001.molmenu-build-model-builddo-optimizationquery-value total-energyfile-format zmtwrite-file 000A-0001.zmtomsgs-not-to-fileexit-script附: HyperChem State Variables (HSV)abinitio-buffer-size: Variable, Read/Write.Type: integer in range (1 .. 2147483647).Two electron integral buffer size.abinitio-calculate-gradient: Variable, Read/Write.Type:boolean.Enable Ab Initio gradient calculation (Single Point only).abinitio-cutoff: Variable, Read/Write.Type: float in range (0 .. 1e+010).Two electron integral cutoff.abinitio-d-orbitals: Variable, Read/Write.boolean.Type:Either five (False) or six (True).abinitio-direct-scf: Variable, Read/Write.boolean.Type:Enable Ab Initio Direct SCF calculation.abinitio-f-orbitals: Variable, Read/Write.boolean.Type:Either seven (False) or ten (True).abinitio-integral-format: Variable, Read/Write.Type: enum(raffenetti, regular).Either regular or raffenetti.abinitio-integral-path: Variable, Read/Write.string.Type:Path for storing integrals.abinitio-mo-initial-guess: Variable, Read/Write.Type: enum(core-hamiltonian, projected-huckel, projected-cndo, projected-indo).Either core-hamiltonian, projected-huckel, projected-cndo, projected-indo. abinitio-mp2-correlation-energy: Variable, Read/Write.boolean.Type:Enable Ab Initio MP2 correlation energy.abinitio-mp2-frozen-core: Variable, Read/Write.boolean.Type:Enable Ab Initio MP2 frozen core.abinitio-scf-convergence: Variable, Read/Write.Type: float in range (0 .. 100).SCF Convergence for Ab Initio.abinitio-use-ghost-atoms: Variable, Read/Write.boolean.Type:Include or ignore ghost atoms.accelerate-scf-convergence: Variable, Read/Write.Type:boolean.Whether to use DIIS procedure.add-amino-acid: Command.Arg list: string.String-1 gives the name of an amino acid residue to add to the system.add-nucleic-acid: Command.Arg list: string.String-1 names the nucleotide to add to the current system.align-molecule: Command.Arg list: .Align the inertial axes of the molecular system.align-viewer: Command.Arg list: enum(x, y, z, line).Align the viewer's line-of-sight with the indicated axis or LINE.allow-ions: Variable, Read/Write.Type:boolean.Whether to allow excess valence on atoms.alpha-orbital-occupancy: Variable, Read/Write.Type: vector of float.(i) Number of electrons in the i-th MO.alpha-scf-eigenvector: Variable, Read/Write.Type: vector of float-list.(i) Coefficients for the i-th MO.amino-alpha-helix: Command.Arg list: (void).Subsequent additions of amino acid residues are to use alpha-helix torsions. amino-beta-sheet: Command.Arg list: (void).Subsequent additions of amino acid residues are to use beta-sheet torsions. amino-isomer: Variable, Read/Write.Type: enum(l, d).Whether amino acids are l or d.amino-omega: Variable, Read/Write.Type: float angle in range (-360 .. 360).The Omega amino acid backbone angle.amino-phi: Variable, Read/Write.Type: float angle in range (-360 .. 360).The Phi amino acid backbone angle.amino-psi: Variable, Read/Write.Type: float angle in range (-360 .. 360).The Psi amino acid backbone angle.animate-vibrations: Variable, Read/Write.boolean.Type:Whether or not to animate vibrations.annotation-color: Variable, Read/Write.string.Type:Default color for annotations.annotation-filled: Variable, Read/Write.boolean.Type:The circle and rectangle annotations are filledannotation-layer-hidden: Variable, Read/Write.boolean.Type:The annotation layer is hidden andd only molecule layer shows. annotation-layer-in-front: Variable, Read/Write.boolean.Type:The annotation layer is in front of molecule layer.append-dynamics-average: Command.Arg list: string.Add a named selection to dynamics average gathering.append-dynamics-graph: Command.Arg list: string.Add a named selection to dynamics graph display.append-omsgs-to-file: Command.Arg list: string.String-1 gives the name of a file to which o-msgs are to be appended. assign-basisset: Command.Arg list: string.Assign a basis set to a selection or system.atom-basisset: Variable, Read/Write.Type: array of string.(iat, imol) The basis set of atom iat in molecule imol.atom-charge: Variable, Read/Write.(iat, imol) The charge of atom iat in molecule imol.atom-color: Variable, Read/Write.Type: array of enum(ByElement, Black, Blue, Green, Cyan, Red, Violet, Yellow, White).(iat, imol) The current color of the atom.atom-count: Variable, Readonly.Type: vector of integer.(imol) The number of atoms in molecule imol.atom-extra-basisset: Variable, Read/Write.Type: array of string, float.(iat, imol) The basis set of atom iat in molecule imol.atom-info: Variable, Readonly.(unknown).Type:Funny composite to support backends.atom-label-text: Variable, Readonly.Type: array of string.(iat, imol) RO. The text of the current atom label.atom-labels: Variable, Read/Write.Type: enum(None, Symbol, Name, Number, Type, Charge, Spin, Mass, BasisSet, Chirality, RMSGradient, Custom).Label for atoms.atom-mass: Variable, Read/Write.Type: array of float.(iat, imol) The mass of atom iat in molecule imol.atom-name: Variable, Read/Write.Type: array of string.(iat, imol) The name of atom iat in molecule imol.atom-spin-density-at-nucleus: Variable, Read/Write.Type: array of float.(iat, imol) The electron density of nucleus of atom iat in molecule imol.atom-spin-population: Variable, Read/Write.Type: array of float.(iat, imol) The spin density of atom iat in molecule imol.atom-type: Variable, Read/Write.Type: array of string.(iat, imol) The type of atom iat in molecule imol.atomic-number: Variable, Read/Write.(iat, imol) The atomic number of atom iat in molecule imol.atomic-symbol: Variable, Readonly.Type: array of string.(iat, imol) The element symbol of the atom.auxilliary-basis: Variable, Read/Write.Type: integer in range (0 .. 3).1=A1, 2=A2, 3=P1back-clip: Variable, Read/Write.float.Type:Set back clipping plane.backend-active: Variable, Read/Write.boolean.Type:Whether current channel is an active backend.backend-communications: Variable, Read/Write.Type: enum(Local, Remote).Whether to compute on local or remote host.backend-host-name: Variable, Read/Write.string.Type:The name of remote host for backend communications.backend-process-count: Variable, Read/Write.Type: integer in range (1 .. 32).The number of processes to run.backend-user-id: Variable, Read/Write.string.Type:The user id to use on the remote host for backend communications.backend-user-password: Variable, Read/Write.string.Type:The password for user id to use on the remote host for backend communications. balls-highlighted: Variable, Read/Write.boolean.Type:Balls and Balls-and-Cylinders should be highlighted when shaded.balls-radius-ratio: Variable, Read/Write.Type: float in range (0.001 .. 1).Size of the Balls relative to the maximum value.balls-shaded: Variable, Read/Write.boolean.Type:Balls and Balls-and-Cylinders should be shaded.basisset-count: Variable, Readonly.integer.Type:Number of coefficients required to describe a molecular orbital.bend-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Results from backend computation.beta-orbital-occupancy: Variable, Read/Write.Type: vector of float.(i) Number of electrons in the i-th MO.beta-scf-eigenvector: Variable, Read/Write.Type: vector of float-list.(i) Coefficients for the i-th MO.bond-color: Variable, Read/Write.Type: enum(ByElement, Black, Blue, Green, Cyan, Red, Violet, Yellow, White).The color used for drawing atoms and bonds.bond-spacing-display-ratio: Variable, Read/Write.Type: float in range (0 .. 1).Bond spacing display ratio.builder-enforces-stereo: Variable, Read/Write.boolean.Type:Whether the model builder implicitly enforces any existing stereochemistry. calculation-method: Variable, Read/Write.Type: enum(MolecularMechanics, SemiEmpirical, AbInitio, DFT).Whether molecular mechanics, semi-empirical, or ab initio.cancel-menu: Variable, Read/Write.Type:boolean.Whether the cancel menu is up, or the normal one.cancel-notify: Command.Arg list: string.String-1 names a variable to stop watching.change-stereochem: Command.Arg list: integer, integer.Immediately change the stereochemistry about (iat, imol).change-user-menuitem: Command.Arg list: integer, string, string.Change the text and procedure associated with the specified user MenuItem. chirality: Variable, Read/Write.Type: array of string.(iat, imol) A, R, S, or ?, for achiral, R, S, or unknown chirality. ci-criterion: Variable, Read/Write.Type: enum(Energy, Orbital).One of: energy, orbital.ci-excitation-energy: Variable, Read/Write.Type: float in range (0 .. 10000).When ci-criterion=energy, maximum excitation energy.ci-occupied-orbitals: Variable, Read/Write.Type: integer in range (0 .. 32767).When ci-criterion=orbital, count of occupied orbitals included. ci-state-to-optimize: Variable, Read/Write.Type: integer in range (0 .. 32767).Which CI state to optimize with conjugate directionsci-unoccupied-orbitals: Variable, Read/Write.Type: integer in range (0 .. 32767).When ci-criterion=orbital, count of unoccupied orbitals included. clip-cursor: Variable, Read/Write.Type: float in range (0 .. 1000).Select Z axis clip cursor tool.clip-icon-step: Variable, Read/Write.Type: float in range (0 .. 1000).Select clip step.color-element: Command.Arg list: integer, enum().Element Int-1 gets color String-2 as its default color.color-selection: Command.Arg list: string.String-1 names a color for the current selection.compile-script-file: Command.Arg list: string, string.Compile file string-1, writing result to string-2 configuration: Variable, Read/Write.integer.Type:The current UV configuration of the system.configuration-interaction: Variable, Read/Write.Type: enum(NoCI, SinglyExcited, Microstate).One of: no-ci, singly-excited, microstate.connectivity-in-pdb-file: Variable, Read/Write.Type:boolean.Whether connectivity information is to be included in a PDB file.constrain-bond-angle: Command.Arg list: float angle in range (-360 .. 360).Float-1 gives the angle constraint for the three currently selected atoms. constrain-bond-down: Command.Arg list: integer, integer, integer, integer.Constrain the bond from (iat1, imol1) to (iat2, imo2) to be down.constrain-bond-length: Command.Arg list: float in range (0 .. 100).Float-1 gives the length constraint for the two currently selected atoms. constrain-bond-torsion: Command.Arg list: float angle in range (-360 .. 360).Float-1 gives the torsion constraint for the four currently selected atoms. constrain-bond-up: Command.Arg list: integer, integer, integer, integer.Constrain the bond from (iat1, imol1) to (iat2, imo2) to be up.constrain-change-stereo: Command.Arg list: integer, integer.Constrain atom (iat, imol) to change the current stereochemistry.constrain-drawing: Variable, Read/Write.boolean.Type:Whether to constrain bond lengths and angles to canonicalize drawing of moleculeconstrain-fix-stereo: Command.Arg list: integer, integer.Constrain atom (iat, imol) to enforce the current stereochemistry.constrain-geometry: Command.Arg list: string.String-1 describes the geometry constraint around the currently selected atom. coordinates: Variable, Read/Write.Type: array of float, float, float.(iat, imol) The x, y, and z coordinates of atom iat in molecule imol. coordination: Variable, Readonly.Type: array of integer.(iat, imol) The coordination number for the specified atom.correlation-functional: Variable, Read/Write.Type: enum(None, Perdew86, VWN, LYP, PZ81, PW91, PBE96, HCTH98).Perdew, LYP, etc.cpk-max-double-buffer-atoms: Variable, Read/Write.Type: integer in range (0 .. 2147483647).Maximum number of double buffered atoms in cpk rendering mode. create-atom: Command.Arg list: integer in range (0 .. 103).Create a new atom at the origin with atomic number nAtno.current-file-name: Variable, Readonly.string.Type:The name of the current file.custom-title: Variable, Read/Write.string.Type:Custom Title string, append string to title.cutoff-inner-radius: Variable, Read/Write.Type: float in range (0 .. 1e+010).The distance (in Angstroms) to begin a switched cutoff.cutoff-outer-radius: Variable, Read/Write.Type: float in range (0 .. 1e+010).The distance (in Angstroms) at which nonbonded interactions become zero. cutoff-type: Variable, Read/Write.Type: enum(None, Switched, Shifted).Electrostatic cutoff to apply to molecular mechanics calculations.cycle-atom-stereo: Command.Arg list: integer, integer.Advance the stereo constraint about atom (iat, imol).cycle-bond-stereo: Command.Arg list: integer, integer, integer, integer.Advance the stereo constraint along the bond (iat1, imol1)--(iat2, imol2). cylinders-color-by-element: Variable, Read/Write.boolean.Type:Color Cylinders using element colors.cylinders-width-ratio: Variable, Read/Write.Type: float in range (0 .. 1).Width of the Cylinders relative to the maximum value.d-orbitals-on-second-row: Variable, Read/Write.Type:boolean.Include D orbitals on second row.declare-float: Command.Arg list: string.Declare a new floating-point variable.declare-integer: Command.Arg list: string.Declare a new integer variable.declare-string: Command.Arg list: string.Declare a new string variable.default-element: Variable, Read/Write.Type: integer in range (0 .. 103).The atomic number of the default element for drawing operations. delete-atom: Command.Arg list: integer, integer.Delete the specified atom.delete-file: Command.Arg list: string.filename to be deleted.delete-named-selection: Command.Arg list: string.Remove the named selection String-1 from the list of named selections. delete-selected-atoms: Command.Arg list: (void).Delete the currently selected atoms.dipole-moment: Variable, Read/Write.Type: float in range (-1e+010 .. 1e+010).Dipolemoment.dipole-moment-components: Variable, Read/Write.Type: float, float, float.Dipole moment components.do-langevin-dynamics: Command.Arg list: (void).Perform a Langevin dynamics computation on the system.do-molecular-dynamics: Command.Arg list: (void).Perform a molecular dynamics computation on the system.do-monte-carlo: Command.Arg list: (void).Perform a Monte Carlo computation on the system.do-optimization: Command.Arg list: (void).Perform a structure optimization on the system.do-qm-calculation: Variable, Read/Write.boolean.Type:For single-point QM calculations, whether to re-compute wave function. do-qm-graph: Variable, Read/Write.boolean.Type:For single-point QM calculations, to graph some data.do-qm-isosurface: Variable, Read/Write.boolean.Type:For single-point QM calculations, to generate iso-surface of results.do-single-point: Command.Arg list: (void).Perform a single-point computation on the system.do-vibrational-analysis: Command.Arg list: (void).Perform a vibrational analysis computation on the system.dot-surface-angle: Variable, Read/Write.Type: float angle in range (-90 .. 90).Dot surface angle.double-buffered-display: Variable, Read/Write.boolean.Type:Whether display operations are double-buffered.dynamics-average-period: Variable, Read/Write.Type: integer in range (1 .. 32767).Computation results from dynamics run.dynamics-bath-relaxation-time: Variable, Read/Write.Type: float in range (0 .. 1e+010).Bath relaxation time for dynamics.dynamics-collection-period: Variable, Read/Write.Type: integer in range (1 .. 32767).Dynamics data collection interval.dynamics-constant-temp: Variable, Read/Write.boolean.Type:Whether to keep temperature fixed at dynamics-simulation-temp. dynamics-cool-time: Variable, Read/Write.Type: float in range (0 .. 1e+010).Time taken to change from dynamics-simulation-temp to dynamics-final-temp. dynamics-final-temp: Variable, Read/Write.Type: float in range (0 .. 1e+010).Temperature to cool back to when annealing.dynamics-friction-coefficient: Variable, Read/Write.Type: float in range (0 .. 1000000).Friction coefficient for Langevin dynamics.dynamics-heat-time: Variable, Read/Write.Type: float in range (0 .. 1e+010).Time taken to change from dynamics-starting-temp ->dynamics-simulation-temp.dynamics-info-elapsed-time: Variable, Readonly.Type: float in range (0 .. 1e+010).Elapsed time in dynamics run.dynamics-info-kinetic-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Computation results from dynamics run.dynamics-info-last-update: Variable, Readonly.boolean.Type:Last update from dynamics run.dynamics-info-potential-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Computation results from dynamics run.dynamics-info-temperature: Variable, Readonly.Type: float in range (0 .. 1e+010).Computation results from dynamics run.dynamics-info-total-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Computation results from dynamics run.dynamics-playback: Variable, Read/Write.Type: enum(none, playback, record).Playback a recorded dynamics run.dynamics-playback-end: Variable, Read/Write.Type: integer in range (0 .. 32767).End playback of recorded dynamics run.dynamics-playback-period: Variable, Read/Write.Type: integer in range (1 .. 32767).Dynamics playback interval.dynamics-playback-start: Variable, Read/Write.Type: integer in range (0 .. 32767).Start playback of recorded dynamics run.dynamics-restart: Variable, Read/Write.boolean.Type:Use saved velocities.dynamics-run-time: Variable, Read/Write.Type: float in range (0 .. 1e+010).Total integration time at dynamics-simulation-temp. dynamics-seed: Variable, Read/Write.Type: integer in range (-32768 .. 32767).Seed for dynamics initialization random number generator. dynamics-simulation-temp: Variable, Read/Write.Type: float in range (0 .. 1e+010).High temperature for the dynamics run.dynamics-snapshot-filename: Variable, Read/Write.string.Type:Name file of to store dynamics run.dynamics-snapshot-period: Variable, Read/Write.Type: integer in range (1 .. 32767).Set recording interval of dynamics run.dynamics-starting-temp: Variable, Read/Write.Type: float in range (0 .. 1e+010).Starting temperature for the dynamics run.dynamics-temp-step: Variable, Read/Write.Type: float in range (0 .. 1e+010).Step size (K) by which temperature is changed.error: Variable, Read/Write.string.Type:The current error.errors-are-not-omsgs: Command.Arg list: (void).Specifies that error messages are to appear in message boxes. errors-are-omsgs: Command.Arg list: (void).Specifies that error messages should be treated like o-msgs. estatic-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Results from backend computation.exchange-functional: Variable, Read/Write.Type: enum(None, Hartree-Fock, Slater, Becke88, PW91, Gill96, PBE96, HCTH98, B3-LYP, B3-PW91, EDF1, Becke97).Slater, Becke88, etc.excited-state: Variable, Read/Write.boolean.Type:False for lowest state, true for next-lowest state.execute-client: Command.Arg list: string.Run a client application.execute-hyperchem-client: Command.Arg list: string.Run a client application. App can reliably connect to instance of HyperChem. execute-string: Command.Arg list: string.Execute the string variable as a script.exit-script: Command.Arg list: (void).Exit the current script.explicit-hydrogens: Variable, Read/Write.boolean.Type:Whether hydrogens are to be drawn explicitly.export-dipole: Variable, Read/Write.boolean.Type:Whether or not to export dipole moment data to .EXT file.export-ir: Variable, Read/Write.boolean.Type:Whether or not to export IR data to .EXT file.export-orbitals: Variable, Read/Write.boolean.Type:Whether or not to export orbital data to .EXT file.export-property-file: Command.Arg list: string.Writes properties to the named file.export-uv: Variable, Read/Write.boolean.Type:Whether or not to export UV data to .EXT file.factory-settings: Command.Arg list: (void).Reset chem to its out-of-the-box state.field-direction: Variable, Read/Write.Type: integer in range (1 .. 3).direction (X,Y or Z) of the static electric field applied to the systemfield-strength: Variable, Read/Write.Type: float in range (-1000 .. 1000).strength (a.u.) of the static electric field applied to the systemfile-diff-message: Command.Arg list: string, string, string, string.Compare file1 to file2; if they are the same say string3, else say string4.file-format: Variable, Read/Write.string.Type:The molecule file format.file-needs-saved: Variable, Read/Write.Type:boolean.Whether the current system needs to be saved.formal-charge: Variable, Read/Write.Type: array of integer.(iat, imol) Positive or negative formal charge on atom used by model builder. front-clip: Variable, Read/Write.float.Type:Set front clipping plane.global-inhibit-redisplay: Variable, Readonly.Type:boolean.Whether redisplay of the system is inhibited (readonly)gradient-x: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Molecular gradient in the X directiongradient-y: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Molecular gradient in the Y directiongradient-z: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Molecular gradient in the Z directiongradients: Variable, Read/Write.Type: array of float, float, float.(iat, imol) The x, y, and z gradients of atom iat in molecule imol.graph-beta: Variable, Read/Write.boolean.Type:If true and UHF, graph beta-spin orbitals instead of alpha.graph-contour-increment: Variable, Read/Write.Type: float in range (-1e+010 .. 1e+010).Increment between contour lines.graph-contour-increment-other: Variable, Read/Write.boolean.Type:Whether to use graph-increment-other (true) or use defaults (false).graph-contour-levels: Variable, Read/Write.Type: integer in range (1 .. 32767).The number of contour levels to plot.graph-contour-start: Variable, Read/Write.Type: float in range (-1e+010 .. 1e+010).Value for first contour line.graph-contour-start-other: Variable, Read/Write.boolean.Type:Whether to use graph-contour-start (true) or use defaults (false).graph-data-row: Variable, Readonly.Type: vector of float-list.(i) The values on the i-th row of graph data.graph-data-type: Variable, Read/Write.Type: enum(electrostatic, charge-density, orbital, orbital-squared, spin-density).The type of wavefunction data to plot.graph-horizontal-grid-size: Variable, Read/Write.Type: integer in range (2 .. 8192).Number of data grid points for plotting in the horizontal direction.graph-orbital-offset: Variable, Read/Write.Type: integer in range (0 .. +Inf).Display orbital offset.graph-orbital-selection-type: Variable, Read/Write.Type: enum(lumo-plus, homo-minus, orbital-number).Display orbital type.graph-plane-offset: Variable, Read/Write.Type: float in range (-1e+010 .. 1e+010).Offset along viewer's Z axis of the plane of the data to plot.graph-vertical-grid-size: Variable, Read/Write.Type: integer in range (2 .. 8192).Number of data grid points for plotting in the vertical direction.grid-max-value: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).The isosurface maximum grid value.grid-min-value: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).The isosurface minimum grid value.hbond-energy: Variable, Readonly.Type: float in range (-1e+010 .. 1e+010).Results from backend computation.heat-of-formation: Variable, Read/Write.Type: float in range (-1e+010 .. 1e+010).Heat of formation.help: Command.Arg list: string.Give help on topic String-1.hide-errors: Variable, Read/Write.boolean.Type:Whether to display error messages on the screen (channel specific). hide-messages: Variable, Read/Write.boolean.Type:Whether to display MESSAGE value on the screen.hide-toolbar: Variable, Read/Write.boolean.Type:Command to toggle the toolbar.hide-warnings: Variable, Read/Write.boolean.Type:Whether to display warning messages on the screen (channel specific). huckel-constant: Variable, Read/Write.Type: float in range (0 .. 10).Extended Huckel constant.huckel-scaling-factor: Variable, Read/Write.Type: float in range (0 .. 100000).Extended Huckel scaling factor.huckel-weighted: Variable, Read/Write.boolean.Type:Extended Huckel weighting factor.。
HyperChem基本操作 Setup设置

HyperChem基本操作Setup设置Charge和Spin Multiplicity在Ab initio和Semi-empirical计算的对话框中出现。
Charge:指定额外的净剩电荷。
额外电荷定义当前的分子系统是一个电中性系统,正电系统(阳离子),还是一个负电系统(阴离子)。
Spin Multiplicity:自旋多重度。
闭壳分子的自旋多重度为1(单重态)。
一个自由基,有一个未成对电子,自旋多重度为2(双重态)。
有两个未成对电子的系统,自旋多重度一般为3(三重态)。
然而在某些情况下,例如两个自由基,两个未成对电子也可能产生单态。
State描述系统中价电子的状态。
包括指定分子处于第一激发单态(Next lowest)或者Lowest电子态。
Lowest给定自旋多重度的最低电子态。
它不一定是基态。
Next lowest给定自旋多重度(单重、双重、三重或四重态)的第一电子激发态。
在HyperChem 6.0中,semi-empirical方法通常需要给定多重度的最低能态(lowest)或者次最低能态(next lowest)的计算。
由于偶数个电子的分子没有未成对电子,是闭壳层单态,所以只有最低三重态是有效的,而次最低三重态是无效的。
例如,苯有偶数个电子,并且基态是闭壳层单重态。
我们可以计算基态(最低单重态),第一激发单重态(次最低单重态),或者第一激发三重态(最低三重态)。
也就是说,或者HOMO被两个电子占据,或者一个电子在HOMO,另一个电子在LUMO,产生了激发单重态或者三重态。
对双重态和四重态,只有给定多重度的最低态(lowest)可用。
UHF选项仅允许给定多重度的最低态(lowest)可用。
例如,可以用UHF选项研究苯的最低三重激发态,但是不能用来计算单重激发态。
这是因为HyperChem中的UHF选项不允许任意的轨道占据,也不允许CI计算。
对于RHF选项,可以计算CI波函数。
这个计算由一系列计算得到的RHF轨道开始,或者从最低单态(或双重态),或者从half electron的单态和三态(或双态和四重态)轨道。
HyperChem

分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
谷晓明 物理化学HyperChem
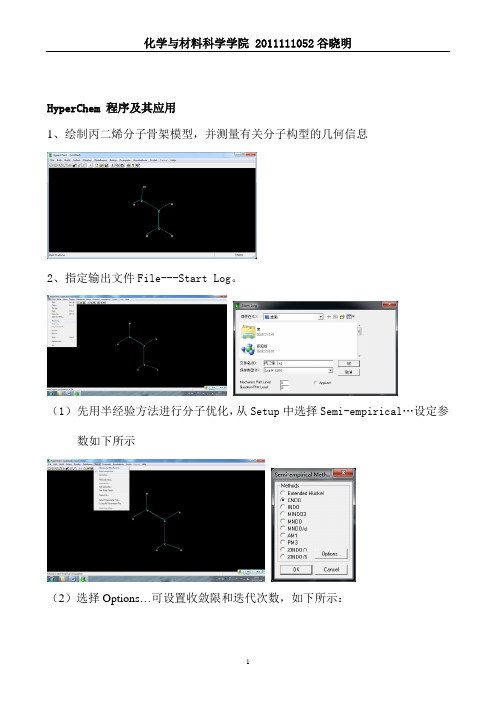
HyperChem 程序及其应用1、绘制丙二烯分子骨架模型,并测量有关分子构型的几何信息2、指定输出文件File---Start Log。
(1)先用半经验方法进行分子优化,从Setup中选择Semi-empirical…设定参数如下所示(2)选择Options…可设置收敛限和迭代次数,如下所示:(3)从Compute中选择Geometry Optimzation…进行集合构型优化:(4)优化完成之后,在Compute选择Single Point可进行单点计算。
3、采用从头算的方法:(1)Setup中选择Ab Initio…设定参数如下:(2)从Compute中选择Geometry Optimzation…进行集合构型优化:(3)完成集合构型优化后,从Compute选择Single Point可进行单点计算。
4、计算结束后,停止数据输出,从File---Stop Log。
5、分析有关分子的性质并简单分析讨论分子性质(1)采用从头算方法后,分析振动光谱:(该图显示谱线的位置、强度和振动模式)虚振动频率-185.84意味着,此结构不是一个稳定结构,而是一个过渡态。
(2)计算电子光谱最低能量跃迁π-π*在373.90,是禁阻跃迁允许的跃迁是116.84单态π-π*跃迁。
(3)分子偶极矩(4)轨道特征1、最高占据轨道2、最低空轨道(5)绘分子图,测电子光谱从Comput选择Plot Molecular Graphs1、2D图像2、3D图像6、结论与经验1、丙烯分子为一平面型分子,并且其振动频率存在虚频-185.84,意味着此平面结构不是一个稳定结构,而是一个过渡态。
2、半经验算法计算分子总能量为-16180.6852898 (kcal/mol),从头算方法计算分子总能量为-72576.4084722 (kcal/mol),所以计算方法的选择很重要。
3、计算分子的电子光谱能够得到该分子最低能量跃迁π-π*在373.90,是禁阻跃迁;允许的跃迁是116.84单态π-π*跃迁。
HyperChem应用-水分子轨道计算

HyperChem应用水分子轨道计算构造水分子1. 在Display菜单,确保Show Hydrogens打开,Rendering对话框Sticks栏中的Perspe ctive关闭。
2. 关闭Default Element对话框中的Explicit Hydrogens,设置缺省元素为O。
3. 在工作区画一个氧原子。
4. 双击Selection激活Model Builder。
它自动为氧原子添加氢原子。
5. 设置Label添加元素符号。
结果是这样:∣H∣∣∣O╲╲╲H键角是109度。
结构校准1. 在Edit菜单选择Align Molecules。
2. 在Align对话框选择Secondary,With对话框选择Y Axis。
3. 关闭Minor Axis。
4. 选择OK。
得到这样的水分子:H H╲╱╲╱╲╱╲╱O5. 把这个结构保存为h2o.hin。
显示原子电荷1. 打开Labels对话框。
2. 把Charge作为Atom的选项,单击OK。
计算波函1. 在Setup菜单选择Semi-empirical。
2. 选择CNDO方法。
选择Options。
当然也可以选择其他方法。
3. 在Semi-empirical的Options的对话框中使用下面的值:Total charge: 0, Spin multiplicity: 1, Spin Pairing: RHF,State: Lowest, Convergence limit: 0.0001, Iteration limit: 50,Accelerate convergence: NO。
这意味着两次叠代计算的值的差小于0.0001 kcal/mol,叠代次数已经达到了最大的次数5 0次。
4. 选择OK回到工作区。
5. 选择Compute菜单中的Single Point。
结果:能量,梯度,和原子电荷如图所示:0.145 0.145╲╱╲╱╲╱╲╱-0.290窗口左下角:Energy=-320.414117, Gradient=124.385845, Symmetry=C2V(结果可能会有略微差别。
HyperChem程序及其应用
河北师范大学计算量子化学研究所蔡新华教授量子化学在线教学.100/qc/lzhx-0.htmHyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作区:状态行:2、打开已存在的数据文件File-Open选择分子图形的显示方式Display- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
Hyperchem学习

OK Thr Mutate
谢谢观赏
胡非非
hyperchem学习 hyperchem
*保存文件
• File→Save As→
保存←
*改变键型连接
• 将鼠标移动至需要修改键型的原子上面,左击鼠标同时移 动至另一个原子上面,则
*制作苯环
• 用鼠标单击环形结构中的任一原子,则可画出苯环
*标记原子
• Display→Labels ↓ →→Symbol→OK ↑ ↓ ↑ ↑ →→↑
*编辑个人原子
Line Options→ 移动鼠标 ← ↓ ←
→Turn on Perspective ↓ ←确定
*缩放工具的使用
按着鼠标左键移 动鼠标
*定心和缩放
• Display Select to Y轴的旋转
*Z轴的旋转
将鼠标移动至工作区, 则可以对分子进行旋转
原子间距离显示在 状态栏
*键角测量
• 按住鼠标左键从碳原子拖到氢原子,接着放开鼠标则
键角显示
六、创建多肽
• • • •
Amino Acids对话框的使用 创建一个多肽 创建一个两性分子 位点专一诱变
*Amino Acids对话框的使用
• Databases→Amino Acids→
不同氨基酸结构
• 双击鼠标左键→Element Table→选择原子→关闭Element Table→将鼠标移动至想要编辑的原子上面,单击左键, 则该原子变为自己后面所选原子,如
*选用模型生成器
• Build→Add H and Model Build→
如果没有显示相应原子,则进入 Display→Show Hydrogens
Hyperchem学习
HyperChem基本操作 单点计算

HyperChem基本操作单点计算单点计算,仅仅执行势能曲面上的一个单个点的计算。
例如对一个双原子分子来说,这可能是在点a原子间距离R=2.0埃的计算。
单点计算的结果给出系统在当前几何构型的势能,以及那个点的梯度。
在点b,c,d,或e的单点计算可能将给出更高的能量。
如果在关键的部分取足够多的点,利用Origin或者Matlab等数学工具软件,就能描出势能曲线,从而精确算出离解能De和核平衡距离re,利用公式就能得到震动参数ωe。
对多原子系统来说,状况更加复杂,但是本质是一样的。
Orbital中显示的轨道信息Alpha & Beta显示选择的轨道是alpha自旋还是beta自旋。
LUMO+显示选择轨道相对于LUMO的位置关系。
例如,选择能量在LUMO之上的轨道,文本框依次显示+1,+2,+3......;如果选择能量低于LUMO的轨道,文本框显示-1,-2,-3......。
HOMO-显示选择轨道相对于HOMO的位置关系。
例如,选择能量在HOMO之上的轨道,文本框依次显示-1,-2,-3......;如果选择能量低于HOMO的轨道,文本框显示+1,+2,+3......。
Number显示从能量最低轨道开始的选择轨道绝对数值。
对于UHF计算,轨道的alpha和beta列编号分开显示,对HOMO-和LUMO+选项也是这样。
Energy以eV为单位显示选择轨道的能量。
Symmetry显示选择轨道的不可约表示。
Labels在轨道显示窗口中,显示每个轨道的电子占据情况和每个轨道的能量。
按照约定俗成,向上的箭头代表alpha自旋,向下的箭头代表beta自旋。
对于RHF计算,最大轨道占据是2;对于UHF计算,最大轨道占据是1。
Zoom Out和轨道的放大如果轨道间距太密,在使用Labels时,文字符号会重合在一起,看起来很不方便。
通过鼠标左键圈出一个或相邻几个选择的轨道,这些轨道就会被放大。
放大之后,Zoom Out选项可以在窗口中重新回到显示全部轨道范围。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用Hyperchem软件进行分子结构构建及性质计算实验目的1.初步了解分子模型方法的原理和应用。
2.学习使用Hyperchem软件构建简单的分子并使用适当方法优化结构。
3.学习使用Hyperchem软件计算简单分子的几何和电子性质。
实验原理化学的学习使我们认识了许多分子的分子式及二维结构,如何得到分子的三维结构,以及分子在空间的几何特征和电子特征,则可以借助于理论计算的工具和方法去模拟计算。
HyperChem软件是HyperCube公司开发的Windows界面程序。
是常用的分子设计和模拟软件。
它可以应用于构建简单及复杂的分子模型并进行综合计算与分析。
分子构建过程可以通过熟练各个菜单及工具栏的操作来实现,计算和分析需要我们了解常用的计算方法。
在本实验室中我们需要了解一下计算方法,这些方法位于HyperChem的Setup菜单下。
(1)分子力学(Molecular Mechanics)方法:分子力学又叫力场方法,目前广泛地用于计算分子的构象和能量。
适用于超大规模体系,超低精度计算。
分子力学的基本假设:玻恩-奥本海默近似,原子核的运动与电子的运动可以看成是独立的;分子是一组靠各种作用力维系在一起的原子集合。
这些原子在空间上若过于靠近,便相互排斥;但又不能远离,否则连接它们的化学键以及由这些键构成的键角等会发生变化,即出现键的拉伸或压缩、键角的扭变等,会引起分子内部应力的增加。
每个真实的分子结构,都是在上述几种作用达到平衡状态的表现。
分子力学从几个主要的典型结构参数和作用力出发来讨论分子结构,即用位能函数来表示当键长、键角、二面角等结构参数以及非键作用等偏离“理想”值时分子能量的变化。
不同的分子力场方法采用不同的势能函数。
MM+:适用于有机分子的计算。
Amber:适用于有机分子、蛋白质和核酸等大分子的计算。
(2)半经验计算(Semi-empirical)方法:是求解HF(Hartree-Fock)方程时采用各种近似,或者直接使用拟合的经验参数来近似求解自洽场。
采用单电子近似,完全不考虑双电子作用而挑选的等效单电子Hamilton算符。
适用于大规模体系,低精度计算。
其中AM1和PM3方法都适用于基态分子的计算。
(3)密度泛函(Density Functional,DFT)方法,是基于量子力学和玻恩-奥本海默绝热近似的从头算方法中的一类解法,是一种研究多电子体系电子结构的量子力学方法。
通过电子密度的泛函计算电子相关。
通过把电子能量分成动能,电子-核相互作用,库仑排斥和说明剩余电子相互作用的交换相关项这几部分进行分别计算。
适用于小规模体系,高精度计算。
DFT中包含很多泛函计算方法,其中B3LYP泛函是经常使用的方法。
此外需要选择合适的基组。
基组是体系轨道的数学描述,对应着体系的波函。
将其带入到薛定谔方程中,就可解出体系的本征值(也就是能量)。
本次实验采用中等基组6-31G(d,p)或6-31G**。
6-31G是劈裂价键基组。
劈裂价键基组可以用于增大所描述轨道的大小(也就是轨道的尺寸),但不能改变形状。
极化函数可以通过给轨道添加角动量来改变轨道的形状(或者说更准确的描述分子的轨道,因为有些分子有很强的共扼体系,对这样体系的描述加极化函数是非常有必要的)。
极化基组6-31G(d,p),表明在6-31G劈裂价键基组的基础上对重原子(如C,N,S等)添加d函数,对H原子添加p函数。
模型计算软件及计算机Hyperchem软件Windows版,电脑一台。
实验步骤构建甲醇,甘油及环己醇分子及性质计算分析(1)打开Hyperchem软件界面,双击工具栏Draw,点中C原子,在界面上单击,出现一个C原子。
(2)点击Build,在下拉菜单里选择Add hydrogens,可以看到界面中出现CH4.(3)双击工具栏Draw,点中O原子,然后在CH4的一个H原子处,点击H原子,这时候H原子就被O原子取代,然后重复(2)操作,可以得到CH3OH.(4)点击Display,在下拉菜单里点Labels,在弹出对话框中选Symbol,可以在界面中看到元素的符号;然后Display--Labels--Bond Length,在界面中出现键的键长,记录下各个键的键长,此为初始键长。
(5)点Select,勾选Atoms和Multiple Selections,然后选中分子中的三个连续的原子,然后点Edit,在下拉菜单里点Set Bond Angle,在弹出对话框里可以看到角度值,记录下这三个原子之间的角度,依次类推,记录其它三个原子之间的键角。
此为初始键角。
(6)点Setup,在下拉菜单里点Molecular Mechanics,在弹出对话框里选MM+,然后OK。
(7)点Compute,在下拉菜单里点Geometry Optimization,然后OK。
可以看到界面窗口中分子在优化过程的变化情况。
计算完成后,我们在界面的左下角会看到Conv=Yes的字样,说明计算完成,还会显示分子的能量E,右下角是选用的计算方法,MM+。
(8)然后Display--Labels--Bond Length,在界面中出现键的键长,记录下各个键的键长。
(9)点Select,勾选Atoms和Multiple Selections,然后选中分子中的三个连续的原子,然后点Edit,在下拉菜单里点Set Bond Angle,在弹出对话框里可以看到角度值,记录下这三个原子之间的角度,依次类推,记录其它三个原子之间的键角。
(10)点Compute,在下拉菜单里点Properties,在弹出对话框里记录Total Energy,Dipole Moment及单位,然后点Details记录下进一步详细的数据。
(11)点Setup,在下拉菜单里点Molecular Mechanics,在弹出对话框里选Amber,然后OK。
(12)点Compute,在下拉菜单里点Geometry Optimization,然后OK。
可以看到界面窗口中分子在优化过程的变化情况。
计算完成后,我们在界面的左下角会看到Conv=Yes的字样,说明计算完成,还会显示分子的能量E,右下角是选用的计算方法,Amber。
然后重复步骤(8),(9),(10),记录相关数据。
(13)点Setup,在下拉菜单里点Semi-empirical,在弹出对话框里选AM1,然后OK。
(14)点Compute,在下拉菜单里点Geometry Optimization,然后OK。
可以看到界面窗口中分子在优化过程的变化情况。
计算完成后,我们在界面的左下角会看到Conv=Yes的字样,说明计算完成,还会显示分子的能量E,右下角是选用的计算方法,AM1。
然后重复步骤(8),(9),(10),记录相关数据。
(15)点Compute,在下拉菜单里点Plot Molecular Graphs,在弹出对话框中Property选Electrostatic Potential,然后在Representation里选2D Contours,这是在界面里出现静电势图,然后点Edit,在下拉菜单里点Copy Image,然后粘贴到Word文档里。
点工具栏里的Select,在界面上点击分子,则图像消失。
这时我们可以来显示其它图形。
同理,点Compute,在下拉菜单里点Plot Molecular Graphs,在弹出对话框中Property选Electrostatic Potential,然后在Representation里选3D Isosurface,然后点Edit,在下拉菜单里点Copy Image,然后粘贴到Word文档里。
(16)点Compute,在下拉菜单里点Orbitals,在弹出对话框中Orbital里选中LUMO (即最低空轨道),在白色长条里输入0,记录Energy和Symmetry,然后Orbital Plotting里选中3D Isosurface,点Plot,界面中出现轨道图,然后点Edit,在下拉菜单里点Copy Image,然后粘贴到Word文档里。
同样,点Compute,在下拉菜单里点Orbitals,在弹出对话框中Orbital里选中HOMO(即最高占据轨道),在白色长条里输入0,记录Energy和Symmetry,然后Orbital Plotting里选中3D Isosurface,点Plot,界面中出现轨道图,然后点Edit,在下拉菜单里点Copy Image,然后粘贴到Word文档里。
计算出(LUMO—HOMO)的能量差值。
(17)点Setup,在下拉菜单里点Semi-empirical,在弹出对话框里选AM1,然后OK。
(18)点Compute,在下拉菜单里点Geometry Optimization,然后OK。
可以看到界面窗口中分子在优化过程的变化情况。
计算完成后,我们在界面的左下角会看到Conv=Yes的字样,说明计算完成,还会显示分子的能量E,右下角是选用的计算方法,PM3。
然后重复步骤(8),(9),(10),记录相关数据。
(19)重复(15),(16)的操作,记录相关数值及复制图形。
(20)点Setup,在下拉菜单里点Density Functional,在弹出对话框里Orbital Basis Set选Large,然后OK。
(21)点Compute,在下拉菜单里点Geometry Optimization,然后OK。
可以看到界面窗口中分子在优化过程的变化情况。
计算完成后,我们在界面的左下角会看到Conv=Yes的字样,说明计算完成,还会显示分子的能量E,右下角是选用的计算方法,Density。
然后重复步骤(8),(9),(10),记录相关数据。
(22)重复(15),(16)的操作,记录相关数值及复制图形。
(23)基于以上对甲醇计算分析的基础上,分别构造甘油和环己醇的分子结构,然后重复步骤(20)和(21)的操作,记录相关数据。
(24)重复(16)的操作,计算出(LUMO—HOMO)的能量差值。
思考题1.比较Hyperchem下各种方法下计算得到的甲醇分子的键长的差别,同时与高斯软件计算的高精度的DFT计算值比较,并计算相对误差。
注:高斯高精度DFT计算值在B3LYP/6-31G(d,p)理论水平下完成。
计算值见下图。
2.比较Hyperchem下甘油和环己醇的DFT计算值与高斯软件计算的高精度的DFT计算值,并计算相对误差。
注:高斯高精度DFT计算值在B3LYP/6-31G(d,p)理论水平下完成。
计算值见下图。
3.简述静电势(Electrostatic Potential)的含义并举例说明分子的静电势可以解释哪些现象。
4.简述LUMO和HOMO,(LUMO—HOMO)能量差值的具体含义。