小木虫 Diamond 软件问题集锦
Materials Studio软件常见问题与解答

目 录Q1:为什么使用Discover进行Dynamics计算时,如果设定了Pressure=1GPa,在计算结果中会出现Pressure等于0,而Stress的XX、YY、ZZ方向为1GPa的情况? (4)Q2:如何在Discover计算中分别对相同环境原子分配不同力场类型? (4)Q3:如何在CASTEP计算中限制某个原子的移动方向? (4)Q4:在安装新的MS时,事先没有停掉License Server,在卸载、安装MS后,发现MS的License Server 无法正常启动。
(5)Q5:如何修改Windows或者Linux下的端口号: (5)Q6:如何使用DMol3进行动力学计算? (6)Q7:如何让Discover程序输出.arc文件? (7)Q8:如何使用rattle关键词来限制水分子的几何结构? (7)Q9,如何使用Standalone方式运行DMol程序? (7)Q10:如何在DMol中加入外界电场? (7)Q12,如何以Standalone方式运行Discover作业? (8)Q13:为什么我在QSAR模块中无法找到新加入的Jurs和DMol3描述符? (8)Q14:如何在DMol模块中,对某一分子只允许其沿着Z方向进行优化,而XY方向则不变? (8)Q15:如果CASTEP计算过程中断电,怎么能够重新开始计算呢?在Keywords中有两个关键词Reuse 和Continuation,它们有什么差异呢? (8)Q16:如果我在Cleave一个平面的时候,选择的是(111)面,或者该晶体原来就是一个三斜晶胞,我怎么才能切出一个长方形的表面来呢? (9)Q17:在使用DMol进行结构优化的时候失败,通过对轨迹的回放发现,整个分子在平面上下进行翻转,并由此导致能量振荡,这种情况应当如何处理? (9)Q18:如何使用XRD数据快速建立相关的晶体结构。
(9)Q19:如何在DMol中考虑溶剂化效应? (10)Q20:如何使用MS软件计算高分子的玻璃化温度? (11)Q21:在使用MS进行计算的时候,中间的xcd文件无法及时更新,而其他文件则能正常显示,为什么? (12)Q22:怎么样在MesoDyn中加入各种不同的限制? (12)Q23:MS给出的DOS和能带图不是很清楚,我能不能自己来做图? (12)Q24:怎么样能让CASTEP在并行计算时更有效? (12)Q25:在使用DMol计算过渡态结构时,经常会发现出现不止一个虚频,怎么回事? (13)Q26:怎么在Altix350上安装MS的Castep和DMol的补丁加速运算速度? (13)Q27:为什么我在Linux下安装license的时候,总是报错呢? (13)Q28:在使用SGI Altix350,打补丁后运行RunCASTEP.sh –np n seedname的时候,出现错误:MPI:asgetnetinfo_array('(null)') failed : array services not available,怎么解决? (14)Q29:MS的GFA是怎么对参数进行杂交的?参数多少与内存有无关系? (14)Q30:DFT方法对计算量和内存的要求是什么样的? (15)Q31:为什么当DMol3在我机器上运行过的时候,总是出现以下错误:floating-point assist fault? (16)Q32:在使用DMol3算频率的时候,突然断电了,怎么才能继续算频率呢? (16)Q33:在Castep模块中,Electronic中的Pseudopotential representation,有Real Space和Reciprocal space,如何取舍? (17)Q34:如何从Discover的输出文件中查看每桢中原子的坐标以及速率等信息? (17)Q35:我使用的是Standalone方式进行Castep和DMol3计算,完成后怎么才能看到最终结构和轨迹呢? (17)Q36:DMol中有TS Conformation也有TS Optimization,应该选取哪一个来搜索过渡态? (17)Q37:在使用CASTEP进行过渡态(TS)搜索时,当用reaction preview产生一个轨迹文件之后,对该轨迹文件进行TS search 运算时,在本机直接计算可以进行,但是进行save files时,却无法完成提示Unable to set UserID. Trajectory file will be invalid. (18)Q38:在Castep中,怎样输出电荷密度?相关数据的单位是什么? (18)Q39:如何在Discover中使用BTCL语言进行多步MD计算? (18)Q40:如何用Dmol3计算Overlay Matrix,并进一步分析? (21)Q41:如何使用Castep程序计算IR振动,为什么计算老是说不收敛? (22)Q42:为什么我在手工使用Castep计算能带结构、态密度或者声子谱的时候,程序始终提示没有Check文件? (22)Q43:Discover的Non-bond中Summation的三种方法有什么区别? (22)Q44:Castep中的Empty Band有何用处?怎么设置? (25)Q45:CASTEP中如何控制能带结构的精细程度? (25)Q46:如何在MS中加入非限制性约束条件,例如约束两个原子间距离? (26)Q47:如何在DMol3中显示大于999号轨道的Homo和Lumo轨道? (27)Q1:为什么使用Discover进行Dynamics计算时,如果设定了Pressure=1GPa,在计算结果中会出现Pressure等于0,而Stress的XX、YY、ZZ方向为1GPa的情况?A1:这是由于在进行Dynamics计算时,选用的Ensemble不相同,如果选用了Parrinello方法,将使用Stress来进行判断,此方法允许晶胞形状与大小都发生变化,已形成剪切,从而使内部的Stress 与外部Pressure相等。
Diamond装机小技巧:Diamond与POV-RAY关联的问题

Diamond 装机小技巧:Diamond 与POV-RAY 关联的问题作者:Monster编辑:CCL 适读人群:正在学习晶体可视化软件的小菜鸟,被Diamond 装机以及与POV-RAY 关联问题困扰的朋友。
最近小编在学习晶体可视化相关知识。
工欲善其事必先利其器,小编选择了较为流行的Diamond 软件。
但在装软件的过程中却遇到了一些问题,最棘手的是如何实现Diamond 与POV-RAY 的关联。
现将这些经验分享出来,希望能够帮到遇到同样问题的朋友们。
1. Diamond 软件安装:本文所用的是Diamond 3 版本,电脑系统为win 10,32 位。
目前,Diamond 已经更新到4.41,官方正式版软件是收费的(可以去度娘找找破J版)。
对于Diamond 3,无需安装得到系统C 盘,直接安装在非系统盘,比如D 盘;新建文件夹,然后按照引导按照即可。
安装完成后在Diamond3/Tutorial 文件夹下打开任意一个实例文件(本教程打开的是C60.cif ,如下图所示)。
届时,Diamond 3 安装完成。
2. 安装POV-RAY:Diamond 安装完成后,软件中已经嵌入好POV-RAY 选项。
依次点击:'Tools' - 'POV-RAY' - 'Render into Bitmap...' ,会弹出如下图所示对话框。
因为你还没有装POV-RAY, 所以Diamond 无法调用POV-RAY 。
POV-RAY 是免费的,直接前往POV-RAY 官网下载即可。
POV-RAY 最新版式POV-RAY 3.7 。
但是POV-RAY3.7之后,Diamond 无法直接调用。
小编我尝试了多种方法都无法实现连用(如果你知道如何解决,或者知道什么原因,欢迎在评论区留言,以方便大家。
)。
无论是安装在Diamond 同一个文件夹下,还是按照在系统C 盘,都无法解决这个问题。
小木虫 Diamond 软件问题集锦

小木虫Diamond软件问题集锦1. Diamond出图时怎么设置dpi数目我在使用Diamond出图时分辨率并不算小,可是编辑说我的图只有96dpi,但杂志要求至少600dpi,请问我该怎么设置? 答: 在picture/layout.../对话框的Targets中选择Bitmap,然后就可以自己输入需要的分辨率了.要保存为位图格式才可以.如果是JPG格式,还是96dpi. 答: 呵呵,发现在tools 下拉菜单的option 里.2. Diamond椭球图阴影画图如图中所示,怎么画出每个椭球中的阴影部分?我用的是3.1d 版本.答: 这个是在diamond中的' 原子设置中' 中选的, 具体如下: picture-------atoms design-------style and colors-------style 中选择octant 即可.3.怎样建立氢键我这儿有一个cif 文件,一个tab 文件和两个已经用diamond作好的图,但不知道这两个图是怎么画出来的,想知道作图过程,请diamond高手用word 或pdf 写出作图过程.答:没什么技术含量的吧,堆积一下,删掉多余的原子,创建氢键就好.4.怎样建立单个分子diamond一打开cif,就是堆积图,怎么才能显示一个分子.答:好像最新的更新包已经改过这个bug 了.但是因为没有对应的破解程序,所以俺还是用以前的 3.0,经常遇到这个问题.如果你的分子间没有作用,即:没有乱七八糟的键连在一起,选中一个原子,然后,ctrol+M, 选中一个分子,然后,反选,即选中除该分子之外的所有分子,然后一键delete, 就ok 了.如果分子连成片,就删吧. 对于非中心分子,即含反离子的分子结构要小心,不要把反离子漏掉了.当然,如果不用画就没问题了.以上拙见,仅供参考. 答:diamond打开cif 时选"建立单个分子",然后点完成,不要点下一步. 请教diamond画C60 遇到的问题(1)我这个C60 在画多面体的时候,中间inset 的atom 的半径在每个面上都要自己一个一个去设定radium,有没有同类原子半径一起设置的?里面没有找到. (2)当我把中间的dummy 原子(就是为了画多面体的时候新加入的)的半径设为0 后,如果想改C60 的每个面的颜色的画,这个怎么改的? (3)最后问一下,我保存的图片中背景的diamond demonstrator 如何去掉. 另献上我第一幅diamond图,如下. 6. 7. 答:1. 可以在atom 设置里找到假原子直接设置 2. 以假原子为中心画多面体 3. 你用的应该是未破解版本. diamond中如何在螺旋链中插入一根棒diamond中如何在螺旋链中插入一根棒,以及如何看结构是单螺旋还是双螺旋结构,如何画出单,双螺旋结构? 答:插入几个哑原子,然后连键,进行一些设置就可以了.前段时间有人传了一些Diamond的说明,其中有一个文件就是详细说明这个棒怎么画的. 找到了这个文件,感谢原作者Crystalsnet,我把它上传到纳米盘,你自己下载下来看看就知道怎么做了. 答:在孔道两侧虚拟两个原子,然后在这两个原子间成建,设置一下键参数就可以了! 答:具体说,选择两端的几个院子,再找这几个的中心位置,分别insert atoms , 再连接心连接, insert bond, 在按你的意思edit the bond. 如何去掉M-C,M-H 连接打开一个晶体数据后,在connectivity 中把M-C,M-H 去掉后.再点complete fragments 第3 页共13 页小木虫Diamond软件问题集锦 8. 9. 10. 11. 12. 13. 后,这些M-C,M-H 又连上了,求助高手. 答:点下connect now 再确定试试. diamond里面如何定义元素化合价怎么样定义元素的化合价,然后使半径比例随之改变??默认设置好像都是原子半径. 答:问题解决了,在atomic parameter 里面. diamond画配位环境时到一定程度原子就长不出来了,是怎么回事今天用diamond画配位环境时画到一定程度(配位环境没画完)原子就长不出来了,不知是怎么回事?请高手指点,谢谢! 答:试试在"Type of sphere"中把Rmax 值设大一些. 答:或者试试用小太阳符号"Fill coordination directly". 修改Diamond的默认单胞数我不知道动了什么参数让Diamond的默认单胞数为 2 了,也就是说每次一打开就是两个单胞里的不对称结构单元,在哪改回去吖? 答:菜单栏的picture-->guidance-->picture creation assistant-->destroy all atoms, bonds 项选勾,下一步-->在fill cell range with 项里选择需要的单胞数,按完成.OK!每次用diamond打开cif 文件时,在引导项中注意选择单胞数,就可以控制显示的单胞数了. 在diamond里怎么让所有一定键长的原子连起来呢如题.我怎么选connectivity,设上DMAX,按OK,不管用呢? 答:我这么做也不管用,后来就在读入cif 之后先不生成图,先在connectivity 里设定要的健长范围就可以了. 答:那是没有设上,该了之后要再点一下需设原子才行. 答:管用的,应该是你操作有问题. 怎么取消透视图我用的是 3.0 读完cif 之后出现的直接是透视图,怎么取消啊? 答:应该是用Representation Settings. 答:用central projection 命令! 绘制黄色和蓝色矩形请高手帮忙看一下图中的黄色和蓝色矩形区域如何画?通过原子建立平面时, 所得的矩形太大,不知道如何调整到所要的大小. 答: 好像Diamond里没法加这样的矩形框, 这个图好像中的矩形框应该是在其它软件中加的,或者不是用Diamond绘制的,比如,可以在Word 里加矩形框,然后设置透明度或许可以. 答:在Diamond里加平面是很大,因为平面是无限延伸的,像这种有限的平面实际是些多面体,通过共面的点建立的,像拓扑图里经常出现一些表示两个面夹角的情况,就是这么做出来的,你可以尝试一下. 答:不错,用加多面体的方法,借助哑原子可以做的. 第 4 页共13 页小木虫Diamond软件问题集锦 14. Diamond结构图寻求完美的颜色对比见识一下各位高手所作的MS 或Diamond做晶体的结构图,主要想参照一下颜色搭配! 答:在polyhedra designs 和atom designs 里面改成你想要的颜色就行了呀. 第5 页共13 页小木虫Diamond软件问题集锦 15. 如何去掉Diamond中画的图Diamond背景字样求助Diamond中画的图copy到word中总有Diamond背景字样,如何去掉呀? 答:你用的那个版本没有破解. 16. 溶剂水没加氢如何画氢键我最近想画个氢键堆积图, 可是溶剂水没加氢, diamond画时溶剂水和羧酸氧上的氢用键显示不出来,求助高手我应该怎么办啊? 答:先加上氢了,或者你把所有的氢都删掉,直接把O-O 之间用虚线连起来就可以了. 答:不加H 直接连接O..O 键容易被审稿人质疑的.最好还是想办法加加H. . 答:给溶剂水加氢,先要从残余峰里面找到和氧的距离比较合适的,然后再去固定住一般就可以的. 答:我都是参照文献的氢键数值,直接在Diamond里设定 D...A 距离然后连接. 实在想做的完美,可以在解析结构时,使用envi X 2,找出原子X 周围较为合理的H, 然后将其固定, 后者先在Diamond里找出形成氢键 D...A 的原子对, 然后在解析结构时在这两个原子之间加入Dummy,并将Dummy 定义为H,然后对H 进行固定,这样可能就合适了. 答:具体的情况很复杂,加H 的技巧很多,实在一言难尽. 17. diamond中怎么画堆积图diamond中怎么画堆积图啊?有机配体要怎么删除啊? 答:啥样的堆积图?把原子半径改大一点不行吗? 答:估计是你填充单胞不够. 18. 原子坐标图中的错误是怎么回事?怎样避免?谢谢. 答: 你那个弹出的错误提示正好把后面的挡住了, 不能移开一点再贴上来吗?只能帮你猜谜了,估计是原子坐标没给,因为其它地方不会提示这个错误的. 答:他好像不是从文件读入的结构信息,而是自己手动输入的,一般绘图软件都可以手动输入的. 19. 如想构造多面体请问如想构造多面体,中心原子有两个如CeVO4,中心原子为Ce 和V 以配位原子为第6 页共13 页小木虫Diamond软件问题集锦 O 来构造两套多面体用什么办法? 答:先选中中心原子,然后在object 里选polyhedra,然后选ligand 做边,如此重复两次就能得到两个多面体. 答:一般XP 都是画配位环境图,基本上所有的图都能用Diamond做. 答:CeVO4 多面体骨架图: 20. 怎样美化diamond图实在没办法,自己对照着说明第一次画图,是LiYF4 晶体. 白钨矿型四方晶系,空间点群C6 4h-I4 1/a,晶胞参数a=5.167,c=10.794 我自己画得不知道是什么,求哪位帮帮忙啊. 第7 页共13 页小木虫Diamond软件问题集锦 21. 22. 23. 24. 25. 26. 答:原子比例不太合适,原子小些棍粗些会比较美观;还有坐标上的箭头最好去掉然后把abc 字体改大点,不然这样的图放到文章上字会看不清楚. 画拓扑重在选择节点,看你选择什么作为结构单元,如果是选某种原子那很方便,在diamond菜单中改变这种原子之间的默认键长; 如果选择的是两个原子的中心或者多个原子的中心,那就需要在shelx 程序中添加原子(cent/x 命令) ,然后再在diamond中改变默认键长.首先自己要了解清楚你的结构,然后选择合适的节点,有的时候从不同角度理解结构,可能会选择不同的节点. 答:画拓扑的话,做好用一下olex,可以自动简化节点. diamond保存图片时可以设置分辨率吗用diamond画图,保存图片选择tif 格式直接保存后,像素很低,感觉不清楚.diamond里可以设置,像素,分辨率,再保存吗? 答:当然可以.在layout 里设置一下,600 最好. 答:一般300 就可以了. 答: "当然可以.在layout 里设置一下,600 最好. "这个好象是对bmp 起作用. 关于diamond中氢键粗线设置问题如题,在diamond中,由build-create h-bond 产生的氢键为较细的虚线,如何把氢键设置得粗一点我试了一下, 如果在picture-bond design 设置粗细, 则氢键也会变成实线, 与其他的化学键分不开.麻烦大家指导如何把氢键设粗又还是保持虚线的形式? 答:首先进入edit,选edit bonds,然后选中你要设置的氢键类型,比如NO键,选中design, 通过调节radius可以调节键的粗细, 然后在fragmenta tion里输入大于1的数字线就成虚线了,我一般选4,你可以自己调节. 答:你可以全选键然后加粗,点应用,不点确定. 控制晶体的层状结构数目Diamond画单晶的结构图,把CIF 文件导入后,用grow 命令的话,晶体的层状结构会一下长很多层,用什么命令才能实现我让它长两层就长两层?我想让它长三层就长三层? 答:用对称操作一个一个加,对称操作码在platon 里面. 答; structure/fill/cell range 里面可以控制大小. 使用diamond生成氢键在使用diamond画图时, craete H-bonds 总是不成功, 没有氢键产生; 实际上存在氢键, 有哪位虫友也遇到过类似问题,帮小弟一把! 答:你设置供体和受体之间的距离了吗?先设置好氢键的距离. 答:必须有氢,否则无法craete H-bonds. Diamond里面如何选择一个分子请问在Diamond里面有没有简单的方法直接选择一个分子?(除了destroy 的时候选中分子中的一个原子就可以删除全部之外) 就像xp 里面的uniq 命令那样,谢谢. 答:右键有一个"select molecule" . 如何用diamond画C-H……pai 和pai……pai 堆积如何用diamond画C-H……pai 和pai……pai 堆积,请多指教. 答:用假原子,可以完成. 答: picture 中的bond designs, 用出现复选框, Atom groups 中选择对象, style 在在and colors 中有一个fragmetation 选项,在它的旁边有一个可供选择的小箭头,按向上的箭头,选择你想要的虚线度,数字越大,虚线的长度越小. 先选定原子,在Diamond(一般是左下角)的picture 工具栏里有一个图标,中间是个第8 页共13 页小木虫Diamond软件问题集锦 27. 28. 29. 30. 31. 棕黄色的小圆球,外面有一圈虚线,按一下那个图标试试看,应该可以. 或者在build 中的fix spere 即可. 或者click built---fill----Rectangular---Area. 怎么插入假原子请教DIAMOND里,怎么在两原子中心插入一个假原子.谢谢! 答:选定两个原子insert 就好啦,呵呵. 如何看两个原子间的距离请问用DIAMOND画图时,如何看两个原子间的距离.期待高手指点. 答:在build 功能里,连及INSERT--bond 两个原子,右键点击即可察看. 答:tool -----measure distance,然后在分别点击你所测的两个原子,将鼠标放到一个原子上就会出现距离. 怎样在diamond里测面与面之间的距离想通过测面面之间的距离来说明π-π 堆积,可是在diamond里面怎样测啊? 答:Diamond的帮助文件里都有详细说明,自己查一下.平行平面之间的距离实际上就是点到面的距离.Diamond可以非常方便的测出,用Tools/Measure Planes etc 命令. 答:先选中一个平面中所需原子, Objects->planes-> creat plane from atoms;同样建立第二个平面.;tools->calculate->distance from planes.Diamond里面原子符号的上标怎么弄想标原子名称,而且有对称符号,如i, ii, iii之类的,请问在Diamond里面怎么把他们弄成上标啊?谢谢! 答:可object--->text 然后打i, 加入后移动到某原子那, 比如Cd1i (i 为上标). 答:要标某原子, 点到那原子, right click, add--->label. 答:画完图后在photoshop 中处理就行. 答:点右键add: atom label;点右键edit atom label 中content 的下拉菜单,找到(individual type) ;然后在下面ndividual 对话中输入你要表达的内容,点OK 就行了. 答:我一般在微软的画图工具中进行标记. 如何用diamond画纳米空心管最近看到了一篇关于纳米管的文献, 很好奇里面的空心管是如何画出来的, 我用diamond画出了那种圆柱管,但不知道怎样把它弄成空心的,因此在这里请教一下高手了. 答:可以参考,photoshop 画空心圆第9 页共13 页小木虫Diamond软件问题集锦 32. 33. 34. 35. 36. 用椭圆选择工具画圆,然后"描边" ,选择好描边的宽度,颜色,位置就可以了.其中, 描边的位置有居内,居中,局外三种,我们把描边宽度设的宽点就可以清楚的看到这三种位置有什么区别. 但是描边的宽度大到一定像素(比如30)就不圆了.这时候的方法是,新建图层上做圆形选区,填充一个颜色,然后变换选区,删掉多余的. 具体做法:Alt+Delete 填充前景色——右键"变换选区"——Shift+Alt+鼠标拖动角部控制块,以中心为固定点等比缩小合适大小——按下Enter 键确定——按下Delete 键删除移动后的选区内容. 变换选区也可以通过按键"Alt+s+t" (Alt+s 是键盘打开"选择"菜单的方式,T是选择菜单下的"变换选区" )实现. 如果使用快捷键"Ctrl+T" ,这是"自由变换"快捷键.它是对被选中的对象进行变换, 不针对选区自身. 如果使用"Ctrl+Alt+T" ,则是对选中对象的复制并进行自由变换.这个当它出现和变换选区一样的手柄框时, 可以用鼠标拖动来看到是复制了一个出来了, 而且是对复制的对象进行变换,也不是对该选区进行变换. 怎样去掉不合理的键有些键是不合理的键连,怎样才能去掉这些键? 答:build--->connectivity 可以改吧. 答:如果你要删除的是一类键,可以在右侧下拉框找到table of bond group,上面会有键的列表,找到你要删的一类键,右键select by group,发现图片上所有的这类键的会被选中.然后再图片上对准选中的其中一个键,右键选删除便可. 答:在buil——connectivity 里,上面是键的类型,下面的框是键长,在上面把你不想要的键的类型前面的框里的对勾去掉,下面的键长可以输入你认为合适的范围. 怎样在grow的时候添加上非键的原子我在用diamond的时候,在使用grow 的时候不知道为什么非键的原子未被增加,请问这个问题怎么解决啊? 答:grow 只会将已经存在的并且成键的原子向外延伸,非键的原子当然就不会增加了. 如果要非键原子一同产生的话可以用fill 功能.diamond画图如何只长几个分子我用diamond画图,是多核的配合物,我想只长出几个分子,怎样可以实现呢,如果把一个晶胞都填充了再删好像很麻烦啊. 答:用GET MOLECULES 命令就可以做到. 答:你的如果不是多核应该很好画的,选中任意一个原子,按住ctrl+alt+S 就可以长出该原子附件的所有原子,叙述不大清楚,楼主试试就可以吧. 答:选中一原子--build---fixed spheres--设置数值. 如何改变背景颜色背景色是黑色怎么变成白色啊? 答:在picture-layout-background 里,也可以刚进去软件时设置. 用diamond输出黑白图因为投稿彩图收费的问题,所以我打算用黑白图画晶体,可是我只会画彩图,不知道怎么弄成黑白的,请高手指点,不胜感激! 答:这个问题很好解决.你将Diamond图片贴到Word 里后,在设置对象格式里将图片颜色设为灰度就可以了. 答:你也可以在用Diamond绘图时就把它绘成灰白的,因为原子的颜色等在Diamond第10 页共13 页小木虫Diamond软件问题集锦里是可以调整的. 37. 用diamond计算MOF微孔得到一个MOF,但不知道怎么算孔有多大? 答:孔隙率还是孔直径?孔隙率的话用PLATON 算,孔直径的话就直接量距离再减范德华半径. 38.diamond的legend问题在diamond中如何把右下角legend中图中没有的原子隐藏起来而只显示图中有的原子. 答:这个没法隐藏的,不过你可以在Structure/Atomic Parameters 对话框里将你不需要的原子删除, 然后再绘图就可以了. 还有一个办法就是做好图后在Build/Atom Groups 对话框中将你计划要隐藏的原子Assign 给其它原子就可以了. 答:可以隐藏,在编辑legend 里面最下面的Select 里,将需要隐藏的原子勾掉就可以了. 答:把鼠标放在legend 上面.点右键,弹出对话框,在display 前的筐子里的钩子去掉即可. 第11 页共13 页小木虫Diamond软件问题集锦 39. 请问怎样在diamond里面看配位数我的理解是这样的,把某目标原子的配位多面体做出来,查顶点个数就行了.但是在diamond里面,做配位多面体的自由度很大,我可以设定配位多面体顶点与目标原子的最大距离,这样一来配位多面体就很主观了,我不知道这个问题怎么办? 答:你应该现在Build/Connectivity 调整键长,然后再做多面体就可以了. 答:这就要依据你的金属原子和配位原子之间的键长了,一般的金属(如主族金属,过渡金属,稀土金属)与常见的配位原子(O 和N)之间的键长都不超过3.0.有的文章即使报道超过3.0 的也不称之为键长, 而是叫做弱作用. 这种弱作用也可以参与多面体的形成.个人意见,仅供参考. 40. Diamond 3 里面如何画示意箭头Diamond 3 里面如何画示意箭头答:我都是先画好图再在PPT 里加的. 答:画好后,转为 .tif 文件后用画图软件添加. 41. Diamond中晶胞的白线如何去除Diamond中晶胞的白线如何去除答:Build/Destroy/All Cell Edges. 答:可以在快截菜单栏点那个X,然后选择cell edge. 42. 求助-如何在diamond中画C-H...π相互作用图急求助高手指点,如何在diamond中画C-H...π相互作用图啊,非常感谢! 答: 你先找到芳香环的中心并给出一个dummy,然后连接H 与dummy, 再把dummy 设置为invisible 就行了. 43. diamond和XP画图及处理图片问题用diamond和Xp画图画完图后,大家用什么方法处理成符合发表要求的图片的?我们是画完图后存成.bmp格式(diamond画图时) ,然后用HyperSnap-DX 5(一种截图软件) 将图片截出来,再直接复制到word文档中,可是这样处理的图像虽然看起来很清楚, 但是打印出来的图像质量很不好(虚) ,请教一下高手,怎么处理才能使打印出来的图像不虚? 答:diamond可以调分辨率的同时我们一般保存为tif 不要bmp 不知道别人如何. 答:直接在pdf 中截图贴进word 也可以啊.要看杂志的,有的杂志要求你提供pdf, 有的是ps,tif,bmp 等不同的. 44. diamond里面的坐标我的diamond里面的坐标看不见了, 我设置好的为什么看不见呢, objects——coordination system,但是还是看不见无论是在边上还是在中心,都看不见,郁闷. 答:在objects-coordination system 里面将X, Y 的值改一下,可能会找到. 第12 页共13 页小木虫Diamond软件问题集锦 45. 讨论一下Diamond ,XP以及Mercury中哪一个画出的图是真实的Diamond ,XP以及Mercury中哪一个画出的图是真实的,这个不太明白啊.大家来讨论一下吧!例如我的一个晶体数据用XP画时,显示高氯酸根中的氧原子与Cu之间有虚线连接(即弱的相互作用) ,而用Mercury看时高氯酸根中的氧原子与Cu则是之间连接了起来,当用Diamond画时单核的Cu则由高氯酸根中的氧原子连接成了一维的链,到底以哪一为准啊? 答:看一下距离吧~和经典键长对比下个人觉得以xp 为准. 答: 主要是设置的键长不同画出来的结构就不同看一下Cu 与O 之间的距离再与经典键长比较看是否成键. 答:diamond的键长也是设置的,在编辑(edit)中就可以随意设置,你所说的问题是diamond默认的. 第13 页共13 页。
界面问题总结

Cu/Diamond界面问题总结(不仅仅是界面问题)一、复合材料热导率(导热性)影响的因素:1.界面热阻(单位面积的界面热阻和界面数)2.致密度解决措施:1.如何降低界面热阻,主要从两个方面:①提高界面结合力:(i)从基体方面考虑:在基体Cu中添加活性元素(Cu与某些元素能够形成共晶合金,如B、Si、C等,要求:1.元素要能够与碳以强键结合形成碳化物。
2.在基体Cu中的分散性好。
)缺点:元素的添加会降低基体金属本身的热物理性能,且不能实现两相界面形貌和成分的直接控制,无法更进一步提升复合材料的热物理性能。
)(ii)从增强体方面考虑:金刚石颗粒表面改性存在的问题:室温下金刚石的热导率虽然高,但高温下的热稳定性差,且易石墨化,金刚石表面会出现裂纹等缺陷;金刚石与一般金属和合金之间有很高的界面能,使金刚石不能被金属和合金润湿,亲和力很差。
方法:金刚石表面金属化,使金刚石具有金属和类金属的性能。
表面化学镀(如盐基型胶体钯活性液的一步活化法镀铜)复合化学镀:Cu-Ni-P、Ni-Cr-P、Ni-W-P等金刚石镀层(研究并不是很多)化学镀+滚镀:可以节约资源和提高镀层质量(对导热性的影响有待研究)优点:镀层在二者之间起结合桥的作用,提高金属对金刚石的粘结能力;金刚石内部缺陷如微裂纹、微小孔洞可通过充填碳化物膜得到弥补,提高金刚石本身导热能力;在复合材料高温烧结时,镀层可以隔离保护金刚石,防止发生石墨化、氧化及其他化学反应。
镀层材料选择的要求:(1).是较强的的碳化物形成元素,可在一定条件下与金属表面发生反应来提高与金刚石的结合力(2).在Cu基体中具有一定的溶解度,可通过扩散来实现其与金属的良好结合,但溶解度不宜过高,以防止基体热导率的恶化(3).镀层本身和其碳化物应具有较高的热导率,避免引入较高的界面热阻存在的问题:以镀Cr为例,由外向里分别是Cu、Cu-Cr固溶体层、Cr、Cr7C3、金刚石。
其中Cu-Cr固溶体层和Cr7C3较薄。
材料课堂——XRD常见问题详解(二),超实用!!

材料课堂——XRD常见问题详解(二),超实用!!展开全文衍射峰的强度和很多因素有关,比如样品的衍射能力,性质,还有仪器功率,测试方法,检测器的灵敏度等等。
XRD 衍射强度和峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关?样品中晶粒越小,衍射峰的峰高强度越来越低,但是峰越来越宽,实际上利用X 射线衍射峰的宽化对样品的结晶颗粒度分析就是根据这个原理的(Scherrer 公式)。
晶粒大小和颗粒大小有关系,但是其各自的含义是有区别的。
一颗晶粒也可能就是一颗颗粒,但是更可能的情况是晶粒抱到一起 , 二次聚集, 成为颗粒。
颗粒不是衍射的基本单位, 但是微小的颗粒能产生散射。
你磨的越细, 散射就越强.。
对于晶粒, 你磨过头了, 晶体结构被破坏了, 磨成非晶, 衍射能力就没有了。
磨得太狠的话,有些峰可能要消失了,而且相邻较近的衍射峰会由于宽化而相互叠加,最终会变成1 个或几个'鼓包'。
一般晶面间距大的峰受晶粒细化的影响会明显一些,因为 d 值大的晶面容易被破坏。
衍射强度变弱本质的原因是由于晶体颗粒变小,还是样品颗粒变小?强度除了和晶粒度有关外,还和晶粒的表面状态有关。
一般颗粒越细,其表面积越大,表面层结构的缺陷总是比较严重的。
结构缺陷将导致衍射强度降低和衍射峰宽化。
XRD 研究的应该是晶粒、晶体的问题,与晶体结构关联的问题,不是样品颗粒的大小问题,谢乐公式算的应该也是晶粒的大小。
样品颗粒的大小要用别的方法测定.,例如光散射、X 射线散射、电镜等。
细针状微晶粉末样品做 XRD 重复性很差。
(制作粉末衍射样品片)怎么可以避免择优取向?择优取向是很难避免的,只能尽力减少他的影响。
首先,你要讲样品磨得尽量细(但要适度,要注意样品的晶体结构不要因研磨过度而受到损坏);不要在光滑的玻璃板上大力压紧(压样时可以在玻璃板上衬一张粗糙的纸张),样品成形尽可能松一些;制样过程中也可以掺一些玻璃粉,或加一些胶钝化一下样品的棱角。
最新Diamond如何找寻经典氢键以及画出

D i a m o n d如何找寻经
典氢键以及画出
如有侵权请联系网站删除,仅供学习交流
找寻氢键的方法:
首先,用shelxtl软件新建项目,用XSHELL打开要找氢键的res文件,删除Q 峰,编辑现在的文件,在命令行输入htab,换行输入CONF,保存文件,删除Q峰,refine,并checek CIF,没问题后,找到LIS文件,查找bonds,找到经典相关信息
画氢键的方法:
打开Daimond 3 按提示打开晶体CIF文件,销毁所有原子,点击build,然后fill,superfill,选择3*3*3,确定;
删除跟氢键无关的氢原子;
点击connectivity设置键长和键角;
点击连接所有原子;
随机选取最长片段并复制;
销毁所有原子并粘贴;
最后精修。
仅供学习交流。
XRD常见问题锦集

X射线衍射常见问题集锦做XRD有什么用途啊,能看出其纯度?还是能看出其中含有某种官能团?X射线照射到物质上将产生散射。
晶态物质对X射线产生的相干散射表现为衍射现象,即入射光束出射时光束没有被发散但方向被改变了而其波长保持不变的现象,这是晶态物质特有的现象。
绝大多数固态物质都是晶态或微晶态或准晶态物质,都能产生X射线衍射。
晶体微观结构的特征是具有周期性的长程的有序结构。
晶体的X射线衍射图是晶体微观结构立体场景的一种物理变换,包含了晶体结构的全部信息。
用少量固体粉末或小块样品便可得到其X射线衍射图。
XRD(X射线衍射)是目前研究晶体结构(如原子或离子及其基团的种类和位置分布,晶胞形状和大小等)最有力的方法。
XRD 特别适用于晶态物质的物相分析。
晶态物质组成元素或基团如不相同或其结构有差异,它们的衍射谱图在衍射峰数目、角度位置、相对强度次序以至衍射峰的形状上就显现出差异。
因此,通过样品的X射线衍射图与已知的晶态物质的X射线衍射谱图的对比分析便可以完成样品物相组成和结构的定性鉴定;通过对样品衍射强度数据的分析计算,可以完成样品物相组成的定量分析;XRD还可以测定材料中晶粒的大小或其排布取向(材料的织构)...等等,应用面十分普遍、广泛。
目前XRD主要适用于无机物,对于有机物应用较少。
关于XRD的应用,在[技术资料]栏目下有介绍更详细的文章,不妨再深入看看。
如何由XRD图谱确定所做的样品是准晶结构?XRD图谱中非晶、准晶和晶体的结构怎么严格区分?三者并无严格明晰的分界。
在衍射仪获得的XRD图谱上,如果样品是较好的"晶态"物质,图谱的特征是有若干或许多个一般是彼此独立的很窄的"尖峰"(其半高度处的2θ宽度在0.1°~0.2°左右,这一宽度可以视为由实验条件决定的晶体衍射峰的"最小宽度")。
如果这些"峰"明显地变宽,则可以判定样品中的晶体的颗粒尺寸将小于300nm,可以称之为"微晶"。
Diamond软件简化节点步骤
基本成型
键的直径设置为等于原子半径
不完整部分
补充细节
切换central和parallel视角
重复以上步骤,选择合适范围得到另一个图
修改分辨率 得到清晰图像
保存
谢谢大家
致谢 感谢 Lotte Hqzhang Crystalsnet 提供做图思路
Grow若干次
删除所有非14.689(2) and 14.746(2) Å的键长
调整到合适的方向
删除所有未成键原子
右
单 键
击
不相键连的各个分子设置成不同的颜色
五重互穿
fivefold interpenetrating
修饰: 删去不规则的atoms 和bonds
Lasso选择:更方便地对结构进行删减
Diamond软件简化节点步骤
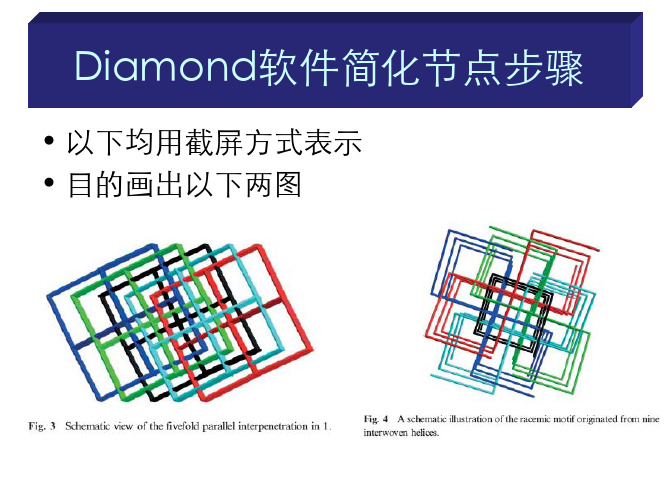
• 以下均用截屏方式表示 • 目的画出以下两图
打开cif文件
Built-Connectivity选择不生长不存在的M-H M-C 同理可以选择其他希望过滤掉的化学键
两个晶体学独立的配体假原子
指定矩形范围添加原子
两个晶体学独立的配体分别作为bi-connector连接相 邻的假原原子 距离为:14.689(2) and 14.746(2) Å
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
小木虫Diamond软件问题集锦1. Diamond出图时怎么设置dpi数目我在使用Diamond出图时分辨率并不算小,可是编辑说我的图只有96dpi,但杂志要求至少600dpi,请问我该怎么设置? 答: 在picture/layout.../对话框的Targets中选择Bitmap,然后就可以自己输入需要的分辨率了.要保存为位图格式才可以.如果是JPG格式,还是96dpi. 答: 呵呵,发现在tools 下拉菜单的option 里.2. Diamond椭球图阴影画图如图中所示,怎么画出每个椭球中的阴影部分?我用的是3.1d 版本.答: 这个是在diamond中的' 原子设置中' 中选的, 具体如下: picture-------atoms design-------style and colors-------style 中选择octant 即可.3.怎样建立氢键我这儿有一个cif 文件,一个tab 文件和两个已经用diamond作好的图,但不知道这两个图是怎么画出来的,想知道作图过程,请diamond高手用word 或pdf 写出作图过程.答:没什么技术含量的吧,堆积一下,删掉多余的原子,创建氢键就好.4.怎样建立单个分子diamond一打开cif,就是堆积图,怎么才能显示一个分子.答:好像最新的更新包已经改过这个bug 了.但是因为没有对应的破解程序,所以俺还是用以前的 3.0,经常遇到这个问题.如果你的分子间没有作用,即:没有乱七八糟的键连在一起,选中一个原子,然后,ctrol+M, 选中一个分子,然后,反选,即选中除该分子之外的所有分子,然后一键delete, 就ok 了.如果分子连成片,就删吧. 对于非中心分子,即含反离子的分子结构要小心,不要把反离子漏掉了.当然,如果不用画就没问题了.以上拙见,仅供参考. 答:diamond打开cif 时选"建立单个分子",然后点完成,不要点下一步. 请教diamond画C60 遇到的问题(1)我这个C60 在画多面体的时候,中间inset 的atom 的半径在每个面上都要自己一个一个去设定radium,有没有同类原子半径一起设置的?里面没有找到. (2)当我把中间的dummy 原子(就是为了画多面体的时候新加入的)的半径设为0 后,如果想改C60 的每个面的颜色的画,这个怎么改的? (3)最后问一下,我保存的图片中背景的diamond demonstrator 如何去掉. 另献上我第一幅diamond图,如下. 6. 7. 答:1. 可以在atom 设置里找到假原子直接设置 2. 以假原子为中心画多面体 3. 你用的应该是未破解版本. diamond中如何在螺旋链中插入一根棒diamond中如何在螺旋链中插入一根棒,以及如何看结构是单螺旋还是双螺旋结构,如何画出单,双螺旋结构? 答:插入几个哑原子,然后连键,进行一些设置就可以了.前段时间有人传了一些Diamond的说明,其中有一个文件就是详细说明这个棒怎么画的. 找到了这个文件,感谢原作者Crystalsnet,我把它上传到纳米盘,你自己下载下来看看就知道怎么做了. 答:在孔道两侧虚拟两个原子,然后在这两个原子间成建,设置一下键参数就可以了! 答:具体说,选择两端的几个院子,再找这几个的中心位置,分别insert atoms , 再连接心连接, insert bond, 在按你的意思edit the bond. 如何去掉M-C,M-H 连接打开一个晶体数据后,在connectivity 中把M-C,M-H 去掉后.再点complete fragments 第3 页共13 页小木虫Diamond软件问题集锦 8. 9. 10. 11. 12. 13. 后,这些M-C,M-H 又连上了,求助高手. 答:点下connect now 再确定试试. diamond里面如何定义元素化合价怎么样定义元素的化合价,然后使半径比例随之改变??默认设置好像都是原子半径. 答:问题解决了,在atomic parameter 里面. diamond画配位环境时到一定程度原子就长不出来了,是怎么回事今天用diamond画配位环境时画到一定程度(配位环境没画完)原子就长不出来了,不知是怎么回事?请高手指点,谢谢! 答:试试在"Type of sphere"中把Rmax 值设大一些. 答:或者试试用小太阳符号"Fill coordination directly". 修改Diamond的默认单胞数我不知道动了什么参数让Diamond的默认单胞数为 2 了,也就是说每次一打开就是两个单胞里的不对称结构单元,在哪改回去吖? 答:菜单栏的picture-->guidance-->picture creation assistant-->destroy all atoms, bonds 项选勾,下一步-->在fill cell range with 项里选择需要的单胞数,按完成.OK!每次用diamond打开cif 文件时,在引导项中注意选择单胞数,就可以控制显示的单胞数了. 在diamond里怎么让所有一定键长的原子连起来呢如题.我怎么选connectivity,设上DMAX,按OK,不管用呢? 答:我这么做也不管用,后来就在读入cif 之后先不生成图,先在connectivity 里设定要的健长范围就可以了. 答:那是没有设上,该了之后要再点一下需设原子才行. 答:管用的,应该是你操作有问题. 怎么取消透视图我用的是 3.0 读完cif 之后出现的直接是透视图,怎么取消啊? 答:应该是用Representation Settings. 答:用central projection 命令! 绘制黄色和蓝色矩形请高手帮忙看一下图中的黄色和蓝色矩形区域如何画?通过原子建立平面时, 所得的矩形太大,不知道如何调整到所要的大小. 答: 好像Diamond里没法加这样的矩形框, 这个图好像中的矩形框应该是在其它软件中加的,或者不是用Diamond绘制的,比如,可以在Word 里加矩形框,然后设置透明度或许可以. 答:在Diamond里加平面是很大,因为平面是无限延伸的,像这种有限的平面实际是些多面体,通过共面的点建立的,像拓扑图里经常出现一些表示两个面夹角的情况,就是这么做出来的,你可以尝试一下. 答:不错,用加多面体的方法,借助哑原子可以做的. 第 4 页共13 页小木虫Diamond软件问题集锦 14. Diamond结构图寻求完美的颜色对比见识一下各位高手所作的MS 或Diamond做晶体的结构图,主要想参照一下颜色搭配! 答:在polyhedra designs 和atom designs 里面改成你想要的颜色就行了呀. 第5 页共13 页小木虫Diamond软件问题集锦 15. 如何去掉Diamond中画的图Diamond背景字样求助Diamond中画的图copy到word中总有Diamond背景字样,如何去掉呀? 答:你用的那个版本没有破解. 16. 溶剂水没加氢如何画氢键我最近想画个氢键堆积图, 可是溶剂水没加氢, diamond画时溶剂水和羧酸氧上的氢用键显示不出来,求助高手我应该怎么办啊? 答:先加上氢了,或者你把所有的氢都删掉,直接把O-O 之间用虚线连起来就可以了. 答:不加H 直接连接O..O 键容易被审稿人质疑的.最好还是想办法加加H. . 答:给溶剂水加氢,先要从残余峰里面找到和氧的距离比较合适的,然后再去固定住一般就可以的. 答:我都是参照文献的氢键数值,直接在Diamond里设定 D...A 距离然后连接. 实在想做的完美,可以在解析结构时,使用envi X 2,找出原子X 周围较为合理的H, 然后将其固定, 后者先在Diamond里找出形成氢键 D...A 的原子对, 然后在解析结构时在这两个原子之间加入Dummy,并将Dummy 定义为H,然后对H 进行固定,这样可能就合适了. 答:具体的情况很复杂,加H 的技巧很多,实在一言难尽. 17. diamond中怎么画堆积图diamond中怎么画堆积图啊?有机配体要怎么删除啊? 答:啥样的堆积图?把原子半径改大一点不行吗? 答:估计是你填充单胞不够. 18. 原子坐标图中的错误是怎么回事?怎样避免?谢谢. 答: 你那个弹出的错误提示正好把后面的挡住了, 不能移开一点再贴上来吗?只能帮你猜谜了,估计是原子坐标没给,因为其它地方不会提示这个错误的. 答:他好像不是从文件读入的结构信息,而是自己手动输入的,一般绘图软件都可以手动输入的. 19. 如想构造多面体请问如想构造多面体,中心原子有两个如CeVO4,中心原子为Ce 和V 以配位原子为第6 页共13 页小木虫Diamond软件问题集锦 O 来构造两套多面体用什么办法? 答:先选中中心原子,然后在object 里选polyhedra,然后选ligand 做边,如此重复两次就能得到两个多面体. 答:一般XP 都是画配位环境图,基本上所有的图都能用Diamond做. 答:CeVO4 多面体骨架图: 20. 怎样美化diamond图实在没办法,自己对照着说明第一次画图,是LiYF4 晶体. 白钨矿型四方晶系,空间点群C6 4h-I4 1/a,晶胞参数a=5.167,c=10.794 我自己画得不知道是什么,求哪位帮帮忙啊. 第7 页共13 页小木虫Diamond软件问题集锦 21. 22. 23. 24. 25. 26. 答:原子比例不太合适,原子小些棍粗些会比较美观;还有坐标上的箭头最好去掉然后把abc 字体改大点,不然这样的图放到文章上字会看不清楚. 画拓扑重在选择节点,看你选择什么作为结构单元,如果是选某种原子那很方便,在diamond菜单中改变这种原子之间的默认键长; 如果选择的是两个原子的中心或者多个原子的中心,那就需要在shelx 程序中添加原子(cent/x 命令) ,然后再在diamond中改变默认键长.首先自己要了解清楚你的结构,然后选择合适的节点,有的时候从不同角度理解结构,可能会选择不同的节点. 答:画拓扑的话,做好用一下olex,可以自动简化节点. diamond保存图片时可以设置分辨率吗用diamond画图,保存图片选择tif 格式直接保存后,像素很低,感觉不清楚.diamond里可以设置,像素,分辨率,再保存吗? 答:当然可以.在layout 里设置一下,600 最好. 答:一般300 就可以了. 答: "当然可以.在layout 里设置一下,600 最好. "这个好象是对bmp 起作用. 关于diamond中氢键粗线设置问题如题,在diamond中,由build-create h-bond 产生的氢键为较细的虚线,如何把氢键设置得粗一点我试了一下, 如果在picture-bond design 设置粗细, 则氢键也会变成实线, 与其他的化学键分不开.麻烦大家指导如何把氢键设粗又还是保持虚线的形式? 答:首先进入edit,选edit bonds,然后选中你要设置的氢键类型,比如NO键,选中design, 通过调节radius可以调节键的粗细, 然后在fragmenta tion里输入大于1的数字线就成虚线了,我一般选4,你可以自己调节. 答:你可以全选键然后加粗,点应用,不点确定. 控制晶体的层状结构数目Diamond画单晶的结构图,把CIF 文件导入后,用grow 命令的话,晶体的层状结构会一下长很多层,用什么命令才能实现我让它长两层就长两层?我想让它长三层就长三层? 答:用对称操作一个一个加,对称操作码在platon 里面. 答; structure/fill/cell range 里面可以控制大小. 使用diamond生成氢键在使用diamond画图时, craete H-bonds 总是不成功, 没有氢键产生; 实际上存在氢键, 有哪位虫友也遇到过类似问题,帮小弟一把! 答:你设置供体和受体之间的距离了吗?先设置好氢键的距离. 答:必须有氢,否则无法craete H-bonds. Diamond里面如何选择一个分子请问在Diamond里面有没有简单的方法直接选择一个分子?(除了destroy 的时候选中分子中的一个原子就可以删除全部之外) 就像xp 里面的uniq 命令那样,谢谢. 答:右键有一个"select molecule" . 如何用diamond画C-H……pai 和pai……pai 堆积如何用diamond画C-H……pai 和pai……pai 堆积,请多指教. 答:用假原子,可以完成. 答: picture 中的bond designs, 用出现复选框, Atom groups 中选择对象, style 在在and colors 中有一个fragmetation 选项,在它的旁边有一个可供选择的小箭头,按向上的箭头,选择你想要的虚线度,数字越大,虚线的长度越小. 先选定原子,在Diamond(一般是左下角)的picture 工具栏里有一个图标,中间是个第8 页共13 页小木虫Diamond软件问题集锦 27. 28. 29. 30. 31. 棕黄色的小圆球,外面有一圈虚线,按一下那个图标试试看,应该可以. 或者在build 中的fix spere 即可. 或者click built---fill----Rectangular---Area. 怎么插入假原子请教DIAMOND里,怎么在两原子中心插入一个假原子.谢谢! 答:选定两个原子insert 就好啦,呵呵. 如何看两个原子间的距离请问用DIAMOND画图时,如何看两个原子间的距离.期待高手指点. 答:在build 功能里,连及INSERT--bond 两个原子,右键点击即可察看. 答:tool -----measure distance,然后在分别点击你所测的两个原子,将鼠标放到一个原子上就会出现距离. 怎样在diamond里测面与面之间的距离想通过测面面之间的距离来说明π-π 堆积,可是在diamond里面怎样测啊? 答:Diamond的帮助文件里都有详细说明,自己查一下.平行平面之间的距离实际上就是点到面的距离.Diamond可以非常方便的测出,用Tools/Measure Planes etc 命令. 答:先选中一个平面中所需原子, Objects->planes-> creat plane from atoms;同样建立第二个平面.;tools->calculate->distance from planes.Diamond里面原子符号的上标怎么弄想标原子名称,而且有对称符号,如i, ii, iii之类的,请问在Diamond里面怎么把他们弄成上标啊?谢谢! 答:可object--->text 然后打i, 加入后移动到某原子那, 比如Cd1i (i 为上标). 答:要标某原子, 点到那原子, right click, add--->label. 答:画完图后在photoshop 中处理就行. 答:点右键add: atom label;点右键edit atom label 中content 的下拉菜单,找到(individual type) ;然后在下面ndividual 对话中输入你要表达的内容,点OK 就行了. 答:我一般在微软的画图工具中进行标记. 如何用diamond画纳米空心管最近看到了一篇关于纳米管的文献, 很好奇里面的空心管是如何画出来的, 我用diamond画出了那种圆柱管,但不知道怎样把它弄成空心的,因此在这里请教一下高手了. 答:可以参考,photoshop 画空心圆第9 页共13 页小木虫Diamond软件问题集锦 32. 33. 34. 35. 36. 用椭圆选择工具画圆,然后"描边" ,选择好描边的宽度,颜色,位置就可以了.其中, 描边的位置有居内,居中,局外三种,我们把描边宽度设的宽点就可以清楚的看到这三种位置有什么区别. 但是描边的宽度大到一定像素(比如30)就不圆了.这时候的方法是,新建图层上做圆形选区,填充一个颜色,然后变换选区,删掉多余的. 具体做法:Alt+Delete 填充前景色——右键"变换选区"——Shift+Alt+鼠标拖动角部控制块,以中心为固定点等比缩小合适大小——按下Enter 键确定——按下Delete 键删除移动后的选区内容. 变换选区也可以通过按键"Alt+s+t" (Alt+s 是键盘打开"选择"菜单的方式,T是选择菜单下的"变换选区" )实现. 如果使用快捷键"Ctrl+T" ,这是"自由变换"快捷键.它是对被选中的对象进行变换, 不针对选区自身. 如果使用"Ctrl+Alt+T" ,则是对选中对象的复制并进行自由变换.这个当它出现和变换选区一样的手柄框时, 可以用鼠标拖动来看到是复制了一个出来了, 而且是对复制的对象进行变换,也不是对该选区进行变换. 怎样去掉不合理的键有些键是不合理的键连,怎样才能去掉这些键? 答:build--->connectivity 可以改吧. 答:如果你要删除的是一类键,可以在右侧下拉框找到table of bond group,上面会有键的列表,找到你要删的一类键,右键select by group,发现图片上所有的这类键的会被选中.然后再图片上对准选中的其中一个键,右键选删除便可. 答:在buil——connectivity 里,上面是键的类型,下面的框是键长,在上面把你不想要的键的类型前面的框里的对勾去掉,下面的键长可以输入你认为合适的范围. 怎样在grow的时候添加上非键的原子我在用diamond的时候,在使用grow 的时候不知道为什么非键的原子未被增加,请问这个问题怎么解决啊? 答:grow 只会将已经存在的并且成键的原子向外延伸,非键的原子当然就不会增加了. 如果要非键原子一同产生的话可以用fill 功能.diamond画图如何只长几个分子我用diamond画图,是多核的配合物,我想只长出几个分子,怎样可以实现呢,如果把一个晶胞都填充了再删好像很麻烦啊. 答:用GET MOLECULES 命令就可以做到. 答:你的如果不是多核应该很好画的,选中任意一个原子,按住ctrl+alt+S 就可以长出该原子附件的所有原子,叙述不大清楚,楼主试试就可以吧. 答:选中一原子--build---fixed spheres--设置数值. 如何改变背景颜色背景色是黑色怎么变成白色啊? 答:在picture-layout-background 里,也可以刚进去软件时设置. 用diamond输出黑白图因为投稿彩图收费的问题,所以我打算用黑白图画晶体,可是我只会画彩图,不知道怎么弄成黑白的,请高手指点,不胜感激! 答:这个问题很好解决.你将Diamond图片贴到Word 里后,在设置对象格式里将图片颜色设为灰度就可以了. 答:你也可以在用Diamond绘图时就把它绘成灰白的,因为原子的颜色等在Diamond第10 页共13 页小木虫Diamond软件问题集锦里是可以调整的. 37. 用diamond计算MOF微孔得到一个MOF,但不知道怎么算孔有多大? 答:孔隙率还是孔直径?孔隙率的话用PLATON 算,孔直径的话就直接量距离再减范德华半径. 38.diamond的legend问题在diamond中如何把右下角legend中图中没有的原子隐藏起来而只显示图中有的原子. 答:这个没法隐藏的,不过你可以在Structure/Atomic Parameters 对话框里将你不需要的原子删除, 然后再绘图就可以了. 还有一个办法就是做好图后在Build/Atom Groups 对话框中将你计划要隐藏的原子Assign 给其它原子就可以了. 答:可以隐藏,在编辑legend 里面最下面的Select 里,将需要隐藏的原子勾掉就可以了. 答:把鼠标放在legend 上面.点右键,弹出对话框,在display 前的筐子里的钩子去掉即可. 第11 页共13 页小木虫Diamond软件问题集锦 39. 请问怎样在diamond里面看配位数我的理解是这样的,把某目标原子的配位多面体做出来,查顶点个数就行了.但是在diamond里面,做配位多面体的自由度很大,我可以设定配位多面体顶点与目标原子的最大距离,这样一来配位多面体就很主观了,我不知道这个问题怎么办? 答:你应该现在Build/Connectivity 调整键长,然后再做多面体就可以了. 答:这就要依据你的金属原子和配位原子之间的键长了,一般的金属(如主族金属,过渡金属,稀土金属)与常见的配位原子(O 和N)之间的键长都不超过3.0.有的文章即使报道超过3.0 的也不称之为键长, 而是叫做弱作用. 这种弱作用也可以参与多面体的形成.个人意见,仅供参考. 40. Diamond 3 里面如何画示意箭头Diamond 3 里面如何画示意箭头答:我都是先画好图再在PPT 里加的. 答:画好后,转为 .tif 文件后用画图软件添加. 41. Diamond中晶胞的白线如何去除Diamond中晶胞的白线如何去除答:Build/Destroy/All Cell Edges. 答:可以在快截菜单栏点那个X,然后选择cell edge. 42. 求助-如何在diamond中画C-H...π相互作用图急求助高手指点,如何在diamond中画C-H...π相互作用图啊,非常感谢! 答: 你先找到芳香环的中心并给出一个dummy,然后连接H 与dummy, 再把dummy 设置为invisible 就行了. 43. diamond和XP画图及处理图片问题用diamond和Xp画图画完图后,大家用什么方法处理成符合发表要求的图片的?我们是画完图后存成.bmp格式(diamond画图时) ,然后用HyperSnap-DX 5(一种截图软件) 将图片截出来,再直接复制到word文档中,可是这样处理的图像虽然看起来很清楚, 但是打印出来的图像质量很不好(虚) ,请教一下高手,怎么处理才能使打印出来的图像不虚? 答:diamond可以调分辨率的同时我们一般保存为tif 不要bmp 不知道别人如何. 答:直接在pdf 中截图贴进word 也可以啊.要看杂志的,有的杂志要求你提供pdf, 有的是ps,tif,bmp 等不同的. 44. diamond里面的坐标我的diamond里面的坐标看不见了, 我设置好的为什么看不见呢, objects——coordination system,但是还是看不见无论是在边上还是在中心,都看不见,郁闷. 答:在objects-coordination system 里面将X, Y 的值改一下,可能会找到. 第12 页共13 页小木虫Diamond软件问题集锦 45. 讨论一下Diamond ,XP以及Mercury中哪一个画出的图是真实的Diamond ,XP以及Mercury中哪一个画出的图是真实的,这个不太明白啊.大家来讨论一下吧!例如我的一个晶体数据用XP画时,显示高氯酸根中的氧原子与Cu之间有虚线连接(即弱的相互作用) ,而用Mercury看时高氯酸根中的氧原子与Cu则是之间连接了起来,当用Diamond画时单核的Cu则由高氯酸根中的氧原子连接成了一维的链,到底以哪一为准啊? 答:看一下距离吧~和经典键长对比下个人觉得以xp 为准. 答: 主要是设置的键长不同画出来的结构就不同看一下Cu 与O 之间的距离再与经典键长比较看是否成键. 答:diamond的键长也是设置的,在编辑(edit)中就可以随意设置,你所说的问题是diamond默认的. 第13 页共13 页。