溶剂效应图解
亲核取代反应中的溶剂效应汇总

a.过渡态电荷分散,形成的偶极—偶极健变弱
b.离去基团为中性,溶剂化程度下降
2 .溶剂对SN2反映的影响
有带电荷的反应物的SN2反应 Nu- + R-L →{Nu&-…R…l&-}#→ Nu—R + LNu: + R-L+ →{Nu&+…R…l&+}#→ Nu+—R + L Nu- + R-L+ →{Nu&-…R…l&+}#→ Nu—R + L (5) (6) (7)
二.亲核取代反应的溶剂效应
亲核取代反应的一般表达式为:
RX + Nu → RNu + X:
Ingold提出:
1. SN1机理
2. SN2机理 亲核取代反应的过渡态一般是偶极型过渡态。 Houghes-Ingold规则:通过过渡态理论来处理溶 剂对亲核取代反应的影响。
1.溶剂对SN1反应的影响
chohchnaohchonachclhcloch反应nu偶极健极性越强过渡态越稳定起始反应物变使反应速度减慢起始反应物变为过渡态时电荷密变化很小或无变化的反应溶剂极性的改变对反应速度无明显形响
亲核取代反应的溶剂效应
任理维
一.溶剂
1. 溶剂的作用
溶解反应物
能与反应物发生相互作用。有机反应中影响到
a.过渡态电荷被分散,形成的偶极—偶极健变弱
b.反应物极性大,溶剂化效应不利于过渡态的形成
例外:a :溶剂化(极性:CH3CH2OH∠H2O)
b: 解离度 (解离:CH3CH2OH∠H2O)
ONa
OCH2 CH2N(CH2CH2)2
OH
溶剂的拉平效应与区分效应

溶剂的拉平效应与区分效应
溶剂是化学反应中不可或缺的一部分,它可以影响反应的速率、选择性和产物的性质等。
在溶液中,溶剂分子与反应物或产物分子之间发生相互作用,这种相互作用可以分为拉平效应和区分效应。
一、拉平效应
拉平效应是指溶剂分子对反应物或产物分子的空间取向进行调整,使得它们更容易相互接触和反应。
这种效应通常发生在极性溶剂中,因为极性溶剂分子具有极性键和极性分子中心,可以与反应物或产物分子中的极性键或极性分子中心形成氢键、离子键或范德华力等相互作用。
例如,在水中,氨分子可以与水分子形成氢键,使得氨分子的氢原子朝向水分子的氧原子,氨分子的氮原子朝向水分子的氢原子,从而使得氨分子的空间取向更加有序,更容易与其他反应物或产物分子发生反应。
二、区分效应
区分效应是指溶剂分子对反应物或产物分子的化学性质进行调整,使得它们在反应中表现出不同的活性和选择性。
这种效应通常发生在非极性溶剂中,因为非极性溶剂分子不具有极性键和极性分子中心,不能与反应物或产物分子形成氢键、离子键或范德华力等相互
作用。
例如,在苯中,苯酚和苯胺的活性和选择性有所不同。
苯酚可以与苯分子形成氢键,使得苯酚分子的羟基更容易被质子化,从而表现出更强的酸性。
而苯胺则不能与苯分子形成氢键,使得苯胺分子的氨基更容易被质子化,从而表现出更强的碱性。
溶剂的拉平效应和区分效应是化学反应中不可忽视的因素,它们可以影响反应的速率、选择性和产物的性质等。
因此,在进行化学反应时,需要考虑溶剂的选择和使用条件,以充分利用溶剂的作用,实现反应的最佳效果。
高中化学竞赛 中级无机化学 质子理论 溶剂体系理论及其拉平效应和区分效应(共24张PPT)

(酸)
2)3HCO3- +3H2O → 3H2O + 3CO2 + 3OH- (碱) 3)3H3O+ + 3OH- → 6H2O
1)+2)+3) :
Fe3+ + 3HCO3- → Fe(OH)3↓ + 3CO2 ↑
NH3 中
CH3COOH + NH3 → CH3COO- + NH4+ (酸)
KH + NH3 → K+ + H2↑ + NH2-
2. 溶剂的拉平效应和区分效应
溶剂不能辨别不同酸碱相对强度的效应叫做溶剂的拉平 效应。
溶剂能辨别不同酸碱相对强度的效应叫做溶剂的区分效 应。
在水中为强酸的物质:如HClO4,HBr,HCl,HI 在水中不能存在 不能区分酸强度:把质子转移给H2O,形成H3O+
Ka > 1
pKa < 0
——强度被水“拉平”到水合质子H3O+的强度
NH4+ + NH2-
与水相似
K = 10-33
中和反应:
水:KOH + H3OI → KI + 2H2O
NH3: KNH2 + NH4I → KI + 2NH3
两性反应:
过量OH-
水:Zn2+ + 2OH- → Zn(OH)2 → Zn(OH)42过量NH2-
NH3: Zn2+ + 2NH2- → Zn(NH2)2 → Zn(NH2)42-
① 纯H2SO4
HAc、HNO3也显碱性 CH3CO2H + H2SO4 → CH3CO2H2+ + HSO4HNO3 + 2H2SO4 → NO2+ + H3O+ + 2HSO4-
溶剂概述和溶剂效应

溶剂概述和溶剂效应摘要:对化学反应中溶剂的种类和作用做概述,以及溶剂效应在紫外,荧光,红外,核磁波谱和液相色谱中的作用。
关键词:溶剂溶剂效应吸收光谱液相色谱1,溶剂1.1溶剂的定义溶剂是一种可以溶化固体,液体或气体溶质的液体,继而成为溶液,最常用的溶剂是水。
1.2溶剂的分类溶剂按化学组成分为有机溶剂和无机溶剂有机溶剂是一大类在生活和生产中广泛应用的有机化合物,分子量不大,常温下呈液态。
有机溶剂包括多类物质,如链烷烃、烯烃、醇、醛、胺、酯、醚、酮、芳香烃、氢化烃、萜烯烃、卤代烃、杂环化物、含氮化合物及含硫化合物等等,多数对人体有一定毒性。
(本文主要概述有机溶剂在化学反应以及波谱中的应用)2,溶剂效应2.1溶剂效应的定义溶剂效应是指溶剂对于反应速率,平衡甚至反应机理的影响。
溶剂对化学反应速率常数的影响依赖于溶剂化反应分子和相应溶剂化过渡态的相对稳定性。
2.2溶剂效应在紫外,荧光,红外,核磁中的应用2.2.1溶剂效应在紫外吸收光谱中的应用[5]有机化合物紫外吸收光谱的吸收带波长和吸收强度,与所采用的溶剂有密切关系。
通常,溶剂的极性可以引起谱带形状的变化。
一般在气态或者非极性溶剂(如正己烷)中,尚能观察到振动跃迁的精细结构。
但是改为极性溶剂后,由于溶剂与溶质分子的相互作用增强,使谱带的精细结构变得模糊,以至完全消失成为平滑的吸收谱带。
这一现象称为溶剂效应。
例如,苯酚在正庚烷溶液中显示振动跃迁的精细结构,而在乙醇溶液中,苯酚的吸收带几乎变得平滑的曲线,如图所示2.2.1.1溶剂极性对n→π*跃迁谱带的影响[2]n→π*跃迁的吸收谱带随溶剂的极性的增大而向蓝移。
一般来说,从以环己烷为溶剂改为以乙醇为溶剂,会使该谱带蓝移7nm:如改为以极性更大的水为溶剂,则将蓝移8nm。
增大溶剂的极性会使n→π*跃迁吸收谱带蓝移的原因如下:会发生n→π*跃迁的分子,都含有非键电子。
例如C=O在基态时碳氧键极化成Cδ+=Oδ-,当n电子跃迁到π*分子轨道时,氧的电子转移到碳上,使得羰基的激发态的极性减小,即Cδ+=Oδ-(基态)→C=O(激发态)。
讲课--溶剂效应

溶剂化过程可被定义为在常温下,溶质分子从 真空的一个固定位置移至溶剂中的一个固定位置 的过程。更精微地说,溶剂效应是溶质和溶剂分子 相互吸引的结果,即溶剂分子通过它们和溶质的相 互作用,累积在溶质周围的过程。这个相互作用的 性质和溶质与溶剂的本质有关。描述溶剂效应的 一个关键参数就是溶剂化自由能ΔGsol(solvation free energy),它是将溶质分子从真空移至溶剂中自 由能的变化值。 一般来说,溶剂效应的计算由三部分组成:静 电势、范德华力和孔洞能。
静电势就是讨论溶质分子与溶剂分子静电极化效 应; 范德华力包括溶质分子与溶剂分子间的吸引力 (dispersion)和排斥力(repulsion); 孔洞能是溶质分子要在溶剂分子中形成孔洞所需 要的自由能。 在对溶液体系的研究中,还有一个重要的参数就是 径向分布函数RDF(r),它用来监视溶液中溶质周围 环境结构性质的变化,它的物理意义是对于任意的 分布,在与α原子的距离为r处找到一个β原子的几 率。
其中自洽反应场方法是目前使用较多的考虑溶剂效应的 方法,即将静态理论方法和连续介质溶剂模型结合起来研究 溶液体系。Poisson-Boltzmann方法就是利用求解PB方程来 计算溶质与溶剂分子间的静电极化作用。AMSOL方法采用 Bom模型,进行分子轨道方法计算,来研究溶液体系。超分子 方法认为溶剂分子可能通过氢键或电荷转移作用与溶质分 子紧密结合在一起,然后将这些额外原子包含在一个更大的 但仍是孤立的体系中进行处理。QM/MM方法是把研究体系 设为几个区域,在中心区域进行高精度的量子化学计算(QM), 在周边区域进行半经验或分子力学计算(MM),该方法也是处 理溶液体系的一个较好的方法,分子动力学或Monte Carlo 模拟溶液体系,最适用于研究溶质分子周围溶剂分子的结构 性质,而对溶剂化自由能的计算则没有自洽反应场方法计算 得到的结果精确。
高等有机第二章-溶剂化效应
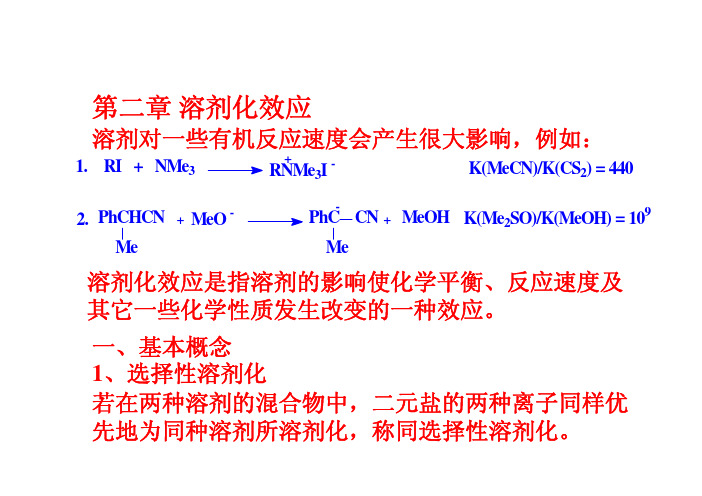
例:CaCl在水-甲醇体系中,Ca2+和Cl–都优先被2水溶剂化。
如阳离子优先被一种溶剂溶剂化,而阴离子优先被另一种溶剂溶剂化,则称异选择性溶剂化。
例:硝酸银在乙腈-水体系中,Ag+优先被乙腈溶剂-优先被水溶剂化。
化, 而NO32、溶剂和溶质分子间的相互作用第一类包括定向诱导力和色散力,这些力是非特异性的,不可能完全饱和。
第二类包括氢键力和电荷转移力,或称电子对授受力。
这类作用有方向并且可以饱和生成化学计量的分子化合物。
C、偶极-诱导偶极力具有永久偶极矩的分子或离子能诱导邻近分子,产生诱导偶极矩,分子在被诱导的瞬间总是处于诱导偶极的方向,两者之间有吸引力。
非极性分子可极化率越大,诱导偶极矩也越大。
这对偶极分子和离子在非极性溶剂中的体系最重要。
D、瞬间偶极-诱导偶极力(色散力〕非极性分子由于电子不断运动,会瞬间产生小的偶极矩,它使邻近分子产生脉冲性极化,从而产生分子间的相互吸引力,这称为色散力。
在两个键已饱和的分子间形成一个附加的成键作用必须是在电子给体分子中存在一个能量足够高的已占据分子轨道,而在电子受体分子中存在一个能量足够底的未占据分子轨道。
3、溶剂的极性和分类(1)质子性溶剂(2)极性非质子性溶剂(3)非极性溶剂量度溶剂极性的标准:(1)偶极矩u常见有机溶剂分子偶极矩的数值在0-5.5D, 在不存在特异性溶质-溶剂间相互作用时,分子极性大小与偶极矩大小一致。
溶剂极性加大,K T 值降低,cis-烯醇式减少。
因为:1、在两种互变异构体中,烯醇式极性较小。
分子内氢键的形成降低羰基偶极-偶极斥力,而在酮式中,这种斥力使其极性加大。
2、分子内氢键使烯醇稳定化,溶剂极性加大,分子间氢键加强,分子内氢键被削弱,烯醇含量减少。
烯醇含量与1,3-二羰基化合物浓度有关。
当偶极性的1,3-二羰基化合物用非极性溶剂稀释,溶剂与羰基作用弱,两羰基偶极斥力大,不稳定;烯醇与溶剂分子间氢键弱,分子内氢键强,烯醇含量增高。
高等有机化学课件电子效应和溶剂效应

对于+I效应也是同样规律, Na(0.9)的电负性小于Mg(1.2) ,前者的活性要大得多,同 样,由于Li的电负性(1.0)很小,给电子能力大于Mg,因此有机锂试剂活性大于格氏试剂, 有很强的亲核能力.
(CH3)3C
O
C(CH3)3
+
1) -78oC (CH3)3CLi 2) H+,H2O
[(CH3)3C]3COH
高等有机化学课件电子效应和溶剂 效应
上述有机化合物分子的酸性变化,都是分子中电子云密度分布发生了变化,或者 从另一个角度说键的极性发生变化的结果。有机分子中,由于电负性不同的取代基 的影响沿着键链传递,致使分子中电子云密度按取代基相对于氢的电负性所决定的 方向而偏移的效应,叫做诱导效应。 (I)诱导效应可表示为:
-I: C CR >-CR=CR >-CR2-CR3 =O>-OR N >=NR>-NR2
CH3COCOOH>CH3OCH2COOH
以上讨论的是都是在静态下分子中电子云密度的分布情况的变化,故称为静态 诱导效应。当某一外来试剂的极性中心接近反应分子时,尤其是在试剂分子开始 进攻而导致过渡态的形成时,也会引起分子中电子云的暂时改变,这种改变叫做 动态诱导效应。这里要注意的是在实际反应中,动态的诱导效应往往起着更重要的 作用,并且有助于反应的进行。幸亏,大多数情况下,静态和动态诱导效应的方向 是一致的。
-I 效应:--NO2>-N+(CH3)3>-CN>-F>-Cl>-Br>-I>-OH
+I 效应: -C(CH3)3>CH2CH3>-CH3 注:这是取代乙酸在水溶液中的离解常数确定的,气相中的结果与此有一些不同
第三章 溶剂效应

+A
-A
B+
A B
A- + B+
A + B+
A B+
在非极性溶剂中有利 在极性溶剂中有利 在极性溶剂中稍有利
B+
B+
+
(4) A+
( 5) A
A B+
在非极性溶剂中稍有利
对溶剂极性不敏感
+ B
A B
A B
反应物
过渡态
产物
如消去反应和亲核取代反应竞争时,溶剂起重要作用。
一般,溶剂的极性大时(如水),容易发生取代反应;
C
C X
同样,由于过渡态的电荷分散程度不同,决定了SN1反 应在极性大的溶剂中进行,E1反应则易于在极性较小 的溶剂中进行。
• 如:三甲基硫正离子的碱性水解速度
(CH3)3S+
+
100oC OH
(CH3)2S + CH3OH
vR H2O 1.0 CH3COOH 19600 • (CH3)3CCl的溶剂分解速度 (CH3)3CCl
5、溶剂化效应的类型:①静电溶剂化效应
②特殊溶剂化效应
6.静电溶剂化效应(靠溶剂的静电作用力) 溶剂化静电理论:用溶剂极性确定相对的溶 剂化能力及其对反应的影响。
(1)溶剂极性对溶质离子化过程的影响;
溶质(R-L)在溶剂S中离子化过程:
R L S [R+L-]
紧密离子对 (A)
[R+ L- ]s
CH 3I + NaCN
CH 3CN + NaI
思考题1.
溶剂: H2O
THF
V = 1.0 V = 5 x 105
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
溶剂效应图解
图解很好!
其实是样品,样品溶剂,流动相和固定相综合作用的关系.当样品在样品溶剂中的相对溶解度大于在流动相时(可以理解为样品溶剂的洗脱能力大于流动相),样品就更喜欢在样品溶剂中,并很想随之流动.但同时与固定相的强作用只能使之形成追赶样品溶剂的效果.最终导致前延峰或裂峰的出现.(如图2:高溶解性溶剂).但当样品与固定相作用很弱时,大部分样品可能会赶上样品溶剂,但又由于与固定相的弱作用,导致其不可能与样品溶剂同时流出,最终导致拖尾峰的出现.
这也就是为什么在一般反相色谱中要用低有机相(比流动相低)溶解样品的原因!其效果就如图1:低溶解性溶剂
样品溶剂效应
很多因素可以导致峰形变差。
样品溶液的组成与进样体积很可能就是导致此种现象的原因。
问题
色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。
此种强溶剂效应的例子在图10-1A中可见。
此处的样品溶液的溶剂是100%乙腈(100%的强溶剂),而流
动相的组成则较弱,18%的乙腈与72%的水。
第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。
当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。
见图10-1B。
解释
当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂(100%ACE),并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉.
当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在图10-2A中, 使用一根短柱,和5UL进样,这与最佳进样体积4UL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如图10-2B),分离度为 2 或更大是评估一个完善方法的一个必要参数,也是每天方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10UL时,分离度更一步降低,此方法就不行了
尽量用流动相去溶解样品,如果样品在流动相中溶解性差不得不用强溶剂溶解,那就尽量减少进样量。