XRD分析软件有4种
XRD经典问题集锦

小木虫XRD问题集锦(第一期)2006 年 10 月 1 日小木虫荣誉出品目录前言 ……………………………………………………………………………………… 问题-:XRD 数据分析讨论 …………………………………………………………… 问题二:双相合金晶胞参数的测定 …………………………………………………… 问题三:很少的粉末样品怎么做粉末XRD …………………………………………… 问题五:XRD拆峰问题如何解决 ……………………………………………………… 问题六:关于XRD知识的问题 ………………………………………………………… 问题八:纳米复合物怎么测XRD ……………………………………………………… 问题九:怎样在origin中处理XRD图………………………………………………… 1 2 8 9 12 13 16 16问题四:不同掺杂金属量的TiO2 看晶形的变化与是否将金属离子掺进去怎么做 … 11问题七:XRD样品相组成分析…………………………………………………………… 13问题十:XRD峰强度问题 ………………………………………………………………… 17 问题十一:样品量少能做XRD粉末衍射吗……………………………………………… 17 问题十二:4 种xrd分析软件功能优劣评比…………………………………………… 问题十四:使用XRD衍射是否可以得到某粉末中的金属含量………………………… 问题十五:如何扣除粉末XRD仪器固有半高宽………………………………………… 问题十六:用XRD测织构,样品如何制备 …………………………………………… 问题十七: 小角度XRD与介孔有序的关系 ……………………………………………… 问题十八:XRD分析时候主要看哪几个指标…………………………………………… 18 20 21 21 22 22 问题十三:用origin做好看的XRD图…………………………………………………… 19问题十九:EDS与XRD的区别 …………………………………………………………… 23 问题二十:xrd的文本导入excle中的问题 …………………………………………… 231小木虫荣誉出品前言通过在小木虫中检索关键词XRD,发现大多数提问及回答都很经典,而 且大多数问题都是我们常见的,为了对小木虫这些珍贵的资源做一保护, 也为了大家查询的方便,特此把大家的提问和回答进行了整理,做此电子 书发表,以后会继续整理此类资源,希望大家满意,多多支持! 由于本人水平有限,如有不足之处敬请各位虫友多多批评指正! 谢谢!特此感谢所有在小木虫中发表XRD贴子及回贴的虫友! 大家辛苦了!编者:rabbit7708 2006 年 10 月 1 日2小木虫荣誉出品问题-:XRD 数据分析讨论 Huangck: XRD 分析是材料分析最基本和最常见的分析技术,我们可以通过它的数据分析出晶 相,晶向,成分,晶粒大小等数据,一般情况,我们借助 PCPDFWIN 和 Jade 辅助分析, 但是一般人分析只是较为浮浅,对软件一些功能的操作还为挖掘出来,大家在分析时对 于数据分析的小技巧,心得讲来分享,分享,让我们也能较好的分析数据. 先找到了两个基本的教程,里面有pdfwin和jade的使用基本的使用方法.台湾国立 成功的PCPDF的pdfwin的教程, (没法上传附件,只好给出连接) /bbs/read.php?tid-40188.html 和国内的Jade数据操作说明 /bbs/viewthread.php?tid=160900 但两个讲的都较肤浅,对于如何分析晶相,晶胞参数,晶粒大小,扣背底等都没有 说明,希望高手能给讲解一下 回答如下: waie_403: 如果用步长为 0.02 度的 x 射线仪扫描得到的 XRD 直接算出来的晶胞参数是不足信 的.好多文章还在煞有其事的讨论晶胞参数的变化,我觉得这一点 是不可取的. Hhxflee: 我现在分析主要用 search-match 结合 pcpdfwin, 当然功能相对简单, 只能扣背底, 剥离 K-aipha2 线. Wangzb: 我认为在保证你的 XRD 数据准确的前提下,应该充分利用 XRD 给我们的信息,包括 角度,强度及峰型方面的信息.大多人只是简单的利用角度方面的信息进行相分析,求 晶胞参数,却忽略了强度方面的丰富信息,而这与你的晶胞内部的原子位置有直接的联 系. Loseman: 其实 jade 用来算晶粒大小并不是非常准确,jade 最强的功能,我感觉是指标化, 然后可以结合 gsas 之类软件进行晶胞参数修正.另外,运用 jade 跟 DB98 结合,计算 混合物的百分含量,也是一个很值得推广的方法.3小木虫荣誉出品Xxxia: 从 XRD 数据的衍射角度分布上我们可以获取晶胞的形状及其晶格参数,而从衍射强 度上我们又可以得到晶胞内的详细信息,比如原子的种类及空间位置,同时也能得到材 料择优取向等信息. Zirconia: 推荐 yuanse 就 jade 使用所做的资料 /showthread.php?t=3183&pp=0 摘取如下(无图片) : NO1 Jade5.0 的安装和设置: Jade5.0 都是自动安装的, 这不成问题. 要把 PDF 卡片引入, 先将 ICDD 的光盘插入, 然 后 pdf/setup/select all/, 其 它 按 提 示 进 行 . 可 以 对 优 选 项 进 行 设 置 : EDIT/preference/,里面包括了对显示窗口的设置,仪器参数的设置,打印输出的设置 等,一般来说按默认就行,我本人则喜欢将 MISC 栏里的"Materials Data, Inc."改 为我自己的大名,哈哈. No2 数据的输入 : Jade 软件可以直接读取 Rigaku,Bruker,Philips,Scintag 等很多衍射仪的原始 数据. 打开 File\patterns,将出现如附件中所示画面, (I) 先 找到你文件位置, (III) 从 的下拉框中选择你的数据格式,按(II)选择.很多仪器输出文件的格式都是*.raw,实 际上都是不一样的,但格式选错了也没关系,软件会给你自动转到合适的格式中去的. 高级一点的:有一些数据格式在(III)的下拉框中没有,比如最常见的 txt,xy 等, 此时你可以自己动手设置,在以上的数据输入面板中,点击工具栏上的"import",进入 格式设置画面,如附件所示,a 区为注释区,b 区为数据格式区,对于最简单的一列角 度,一列强度的数据格式,a 区不用填写,b 区在"angle column"前打上勾,数据从 第 1 行开始读,每行 1 列数据,强度数据从第 8 行开始(角度不算) ,角度从 1 至 6 列, 所得数据格式即为附件中所示的数据格式.你也可以按照自己的数据格式进行自由改 动,如果 a 区中表明第 1 行有说明文字,则数据从第 2 行读入,相应在 b 区就将 data starts 改成 2.做完上面的工作后,将文件后缀改为你的数据后缀(箭头所指) ,再将 该格式保存下来便可大功告成了. No 3 基本功能使用:平滑,扣背底 一张 XRD 图谱出来,往往因为有空气散射,漫散射,荧光以及样品结晶差等等原因 而造成图谱上存在许多"毛刺"和较高的背底,虽然提高 X 光强度能成倍提高信噪比, 然而有时受仪器和样品所限,这两项功能需要用到.但根据我个人的经验,要尽量少使 用平滑和扣背底, 因为这两项操作带来的可能后果就是将一些微弱的有用信息一概抹掉4小木虫荣誉出品了,特别注意的是,如果将数据用来做 Rietveld 精修,更不要进行这两项操作.当然, 如果是将图谱打印出来给别人看,适当进行平滑和扣背底也是个不错的选择. 1 平滑 打开 Filters/smooth pattern 或在快捷工具栏中右键点击也可.随后将出现一个悬浮 框,最上面的一栏中方块可以直接用鼠标拖动,大家试试看图谱会有什么变化,拖到什 么位置,根据情况而定,我的经验是将方块拖到尖峰的底部出现倒生的毛刺之前.再下 面有"parabolic filter"和"quartic filter"的选择,选择后一个的效果稍好.再下 面还有选择框,我一般都不管它.作完以上操作后,再用鼠标左键点击快捷工具栏中的 平滑图标即可. 2 扣背底 打开 analyze/fit background 或在快捷工具栏中右键点击也可.随后也出现一个 如附件中所示的悬浮框, (I)处所示代表了背底拟合的级数,点击越靠前,该级数越高, 也可在右边选择是一次拟合,抑或二次和三次拟合,试情况而定,背底偏离线性越远, 则拟合的级数要求越高. 背底曲线用黄线表示,红点代表了背底在局部的最高点,左键点击图谱上的某一处 便可在此处添加红点,右键点击红点可以消除该点.背底是否拟合好要靠肉眼观察. II 所指为背底以下和以上的面积,可以用来粗略估计样品的结晶度. 以上完成后点击"removal"即可. No 4 寻峰和峰型拟合 1 寻峰 在寻峰之前最好用标样校正过 2theta 值,这些都是仪器操作人员的事情,我就不 说了. 打开 analyze/find peaks,或右键单击快捷工具栏中的图标.出现一个悬浮框,在 "search"里面可以对寻峰的判据进行设置,大家可以改动不同的限制条件,然后按 "apply"看看有什么变化.在"Label"里面主要是可以选定标注的内容,如 d 值,2 theta 值,半峰宽,强度等,这些大家多试几次就 OK 了.如果要将寻峰结果列出,按下 "report"即可. 也可以自己手动直接在图谱上标峰, 大家可以看到有一个悬浮式快捷编辑框"edit toolbar", 左键点击第 3 个图标. 然后就可以直接在峰顶标注, 如果同时按住"Ctrl" 键,则可以在图谱的任何位置上进行标注.如果要去掉某个峰的标注,则用鼠标将竖线 拖到与峰的标注线重叠,出现红颜色之后点击右键即可.如果要删除所有的峰的标注, 在窗口中点击右键,选定"Erase all"即可. 2 峰型拟合 如果要得到精确的峰型,峰位信息,一般都要经过峰型拟合,JADE 提供了单峰拟合5小木虫荣誉出品的功能.在峰型拟合前不要进行平滑和扣背底,也不主张预先自动寻峰. 就像前面一位朋友说的,对于重叠峰的分离比较有难度,也可能会导致软件"罢 工"(我也遇到过这种情况) ,但对于多数人来说,峰型拟合就等同于分峰,因此面对 重叠峰是不可避免的,我的建议是高角度的峰舍弃,并且分峰要一小段接一小段地进行 (背底严重的除外) . 先在图谱上选好角度范围,在编辑栏中左键点击第 7 个图标,然后再右键点击,将 出现一个峰型拟合的工作框(也可通过 analyze/fit peak profile) ,然后选定你的峰 (注意排除 Kalfa2)或者按下"initialize",再按下"refine"即可.有时不管怎样 似 乎 都 拟 合 不 好 , 此 时 可 在 工 作 框 中 调 整 峰 型 函 数 , 以 及 "Exponent" 和 "lorentzian"的值,也可以调整背底的类型等,另外,可以重新确定峰位,去掉一个 计算峰也可以用鼠标的右键.拟合好坏的标准除了看 R 因子外,主要是观察差值线. 按下"report"直接查看结果,里面也有一些设置,如要修正的参数,晶粒大小计算的 根据等,大家在实践中多试试,看看结果会有什么不同.在结果的列表中,选定复选框 可以将该峰定义为背底, 一行被选定 (在图谱上该峰的标注线显示红色) 按下"Erase" , 删除,其它栏目大家都比较熟悉,最后一项为晶粒大小的计算结果,要注意的是,这种 晶粒大小的计算是建立在仪器的实际宽化曲线已存入的基础上的, 否则就按默认的仪器 曲线(软件自带) ,但不同仪器有不同的宽化曲线,因此最好自己建立,这也是仪器操 作人员的事情,我就不多讲了,如果有朋友感兴趣,可以和我直接交流.再后面的事就 是按下"export"进行保存了. 大家都很关注在 Jade 中如何进行晶胞修正的问题,实际上,晶胞修正和指标化是 结合在一起的,因此,如果大家不是对已知晶系很有把握的话,最好在修正之前都预先 进行指标化以判断或确证晶系. 在指标化之前,大家有几项工作需预先进行,一是 2theta 校正(用标样即可) ;二 是相分析,如果是二相甚至多项共存,则归属峰的时候要非常仔细.三是峰型拟合确定 峰位(如果有重叠峰则更加必要) ;我个人认为,指标化最关键的一步是确定峰位,选 取的峰最好是低角度的独立峰,而且选取的峰的数量并非多多益善,只要满足了不同的 晶系要求即可,遇到重叠峰的时候一定要小心! 做好上述工作之后,下一步就是 options/pattern indexing,出现一个面板,在你 的晶系前划勾,然后选择指标化方式,建议是从快(rapid indexing)到慢(exhaustive indexing) ,再下面有 2 theat error 一栏,如实填上即可,晶胞参数最大值最好填的 大一点,其它的按默认,另外,对于一些做有机大分子晶体的朋友来说,晶胞密度可能 比较重要,那就要把 Z 值和化学式输入,根据晶胞体积软件会自动给出的. 完成上述步骤之后,就"go"吧,软件会按照一定的排列方式将很多种可能的结果 一一列出来,fm 值越小,结果的可信度就越高,有时会发现你认为正肯定确的空间群没6小木虫荣誉出品有出现在结果中,不要紧,在"space group"旁边有一个'?',点击它一些,看看有没有 找到,晶体学上,很多空间群都统属于同一点群,因此在指标化时,它们都是等同的. 当你选取其中的一个指标化结果时, 相应的就会在图谱上出现一系列黄色的指标化的衍 射峰位,你可以一一与实际的衍射位对应,看看结果是否准确.有时一个结果也没有得 到,有可能的原因是选取得峰不对,2theta 偏差不对,选择的晶系不对,也有可能最大 晶胞值太小,或者 fm 截取值太低的缘故.那就重新再来吧.接下来的事情就比较容易 了,先选定正确的结果(蓝) ,点击"Refine",出现"Cell Refinement"的控制面板, 再点击 Refine,你就可以得到精确的晶胞参数,esd 表示的是标准偏差. 如果你已知了空间群,并且选峰正确(手动) ,那么你也可以直接"options"/"Cell Refinement"/Refine,打开'reflections',看看偏差多大,它可以作为指标化和晶 胞修正的结果输出. NO.5 物相鉴定 我以前用过专门的 PDF 软件做物相鉴定,但是感觉比较费神,最重要的是不能导入 实验的衍射谱,除三强线外的其它谱线要靠人工去和标准谱一一对应,给准确判定带来 不少困难.JADE 在这方面还是蛮出色的,只需轻点鼠标,很快就能出结果,并且非常直 观, 易于判别, 这主要得益于它具有强大的图谱处理综合功能以及先进的图谱鉴定方法, 在此不作多讲,主要说说如何具体运用. 在进行物相鉴定之前,最好尽量多地知道样品的元素信息,有很多朋友在作定性分 析之前,总是不愿意告知元素组成,甚至有人要用 XRD 去判别元素,这些都是对 XRD 物 相鉴定的一种曲解.XRD 的作用对象只能是"相",而且是"结晶相".同一种结构, 往往它的化学组成形式可以有成百上千种, 而同一个化学组成, 它也可以存在多种结构, 前一个问题的解决主要靠化学分析,后一个问题的解决则非 XRD 莫属(TEM 也能,但不 普遍) . 在进行物相鉴定之前, 预先作背底扣除, 最好不平滑. 右键点击工具栏上的"S/M", 出现一设置面板,"General"里面首先是样品类别的选定,一般而言,我都将类选得 大一些,以防不测.除了物性类别外,其它的还有诸如计算谱,结构被无机晶体结构数 据库收录的图谱等.再下面是一些复选框,如单相鉴定,有否择优取向,是否自动匹配, 以及化学元素限定等等.最后一项是最常用到的,复选"use Chemistry",点击 "Chemistry",出现元素周期表,直接点击你确定的元素,一次为"可能"有,二次 为"肯定"有,建议点击一次变为蓝色即可,然后"OK".除 Chemistry 外,其它的诸 如 Formula,Unit Cell 等等也可以,但都不常用.对于一般的用户来说,完成上述操 作即能进行下一步的工作. 点击设置面板上的"OK",将出现检索结果,一般默认按可信度排列,点击名称, 在图谱上就会出现对应的谱线,在复选框内打勾,就表示选定.到此为止,基本的操作7小木虫荣誉出品就算完成. 幻影无痕: 我觉得去参考一下这本书比较好: 现代物理测试技术. 作者: 梁志德. 出版机构: , , 冶金工业出版社. ISBN :, 7-5024-3186-1.书中对XRD做了专门的介绍! /book_detail.asp?bookid=486 jinbo8125: 清华材料论坛那里的讨论更加深入一些,可以到那边去学 Cntristone: 我测过一种晶体的 xRD 结果 发现 不同的强度 谢 Zirconia: 若是同一个样品 XRD 强度不重复,在仪器正常的情况下,说明可能有择优取向(尤 其片状,针棒状极易发生) ,可加入非晶物质如淀粉予以消弱 这说明了什么呢 请各位指点,谢问题二:双相合金晶胞参数的测定 Buaahao: 我做了一个双相合金的 xrd.这个合金的晶胞参数怎么算呢?双相合金中,一种为 hcp, 一种为 bcc.我想得到 hcp 相的参数,是不是只要把 hcp 相的衍射峰标定后,计算就可以了? 另外大家处理相的晶胞参数的时候,都使用的是 average lattice constants,还是根据 某个晶面算出的参数?哪个更可靠呢?晶面的选择有要求么? 谢谢? 回答如下: Whulsz: 我以前试过,同一个样品如果 x 光测试的位置不同,那么 XRD 的数据还是有很大的 差别. Baosan 俺是个新手,大家多多讨论呀.以前测过 xrd 的数据,同楼上的说得差不多,不同8小木虫荣誉出品区域,不同机器测的峰强都不一样,所以就没有考虑用峰强来做深入的分析. lzg321: jade 好像在标定未知晶体结构的花样时,好像会有不少的难度! xingzhu10755: 深有感触,感觉现在用的软件分析进行科学探讨误差太大了 Punkx: 一般用 xrd 测量晶粒尺寸小于 100 纳米,若是大于 100nm 的晶粒尺寸怎么计算,根 据 XRD 数据. Liangzaili: highscore 不是更好吗? Wustliang: 我是做粉末衍射样品的, 在这里只就粉末衍射说一点看法. pcpdfwin 的功能比较单 一,如果想做的精细就要求从一开始样品的制备与众不同. 首先粉末衍射要求样品的粒度要在 330-450 目,当然理想状态是 1um,但在实际上 很难做到,即使研磨到 1um 了,晶格也破坏的差不多了. 其次调整衍射仪,一般放个多晶 Si 片就可以了,检查仪器的状态.当然在实际测 量的时候最好是在样品里加入内标相,这样很多影响因素就不用一一校正了. 扫描的步径不是越小越好,一般 0.01-0.005 也就可以了.因为做结构精修时,要 求步径为最小 FWHM 的 1/10 也就足够了.最强计数要在 10000-20000 之间. 问题三:很少的粉末样品怎么做粉末XRD Iceray 粉末样品,苦于量奇少,怎么做粉末 XRD?我试过在玻片上沉积一层样品的方法. 但出来的是玻璃的散射, 根本没峰. 在这个帖"求助: XRD 测织构, 用 样品如何制备呢? 作者: gqken 发布日期: 2006-6-23"上看过 有说用样品分散到乙醇中,滴到上面做 的, 但是这种方法能用在粉末衍射上吗?我这里的 XRD 是 BRUCKER 的, 性能一般. 请问, 有什么更好的办法吗?或者有人试过分散到乙醇中的方法吗,效果怎么样 回答如下:9小木虫荣誉出品cad_0: 看有多少啦,一般只要够堆积超过槽的上平面就可以了.不用全部扑满的,只要能 在中间堆积个小山,那玻片一压就可作了. Jiazang: 如果条件允许,不妨自己做一个样品架,然后把衍射仪的狭缝调小一些,这样可能 能够解决你的问题! Iceray: 少到几乎就没有的地步,我是用气相扩散做的,大概能有零点零几克.对小山的方 法根本不够用.衍射是系里公用的.我不太可能调仪器参数.我主要想问有朋友用过乙 醇分散的方法吗 sima022225: 用过,不过效果我用的时候不是很好,做出来晶相不好(我是做 TiO2 的) . Hqzhang: 能做,我知道现在的单晶衍射仪就可以做.虽然不是单晶,但单晶衍射仪安了附件 的话可以测 xrd.我们系的就可以也用不了多少样品. Iceray: TiO2 的峰本来就有点复杂.你做得是粉末衍射吗.你们的仪器是啥型号的 我做得是 CaCO3.峰很简单的 Jiazang: LZ 能否考虑电子衍射? cad_0: 那就再作一些样品吧,我这里不够,就又做了不少.样品一定要够,要不是后期的 性质就不好作了. wanwei006: 喷在铜网上试试看10小木虫荣誉出品问题四:不同掺杂金属量的TiO2,看晶形的变化与是否将金属离子掺进去,怎么做 哲舟: 我作出了掺金属的TiO2,现在有不同掺杂量的样品,也有在最佳浓度时不同灼绕温度的 样品,我要做XRD,看看晶形的变化与是否将金属离子掺进去了,因为我降解污染物的结 果是500度的与掺铁量为0.05%的最好,用检测来证明,上面两组样品,选哪个 好啊?有什么区别呢?是不是都要做的必要啊? 回答如下: Juloong: Quote: 看看晶形的变化与是否将金属离子掺进去了 这个必须至少选三组样品吧. 1,没有掺杂的;2,最佳掺杂;3,过量掺杂 Quote: 最佳浓度时不同灼绕温度 也至少要选三组吧: 1,低于最佳温度;2,最佳温度;3 高于最佳温度 当然,如果感觉三组不够充分的话,你可以多测几组 Juloong: 做 XRD 需要样品的量很少,一药匙已经不少了 Noontgc: 如果是简单看一下,有没有掺杂进去,把哪个 0.05 的样品做一下 xrd, 看有没有掺 杂物质的峰,如果没有,就可能掺杂进去了,如果有掺杂物质的峰出现,有两种情况: 1 是没有掺杂进去, 2 是掺进去少量. 如果第二种情况出现,这时候可以对第二种情况进行详细的分析.再做几个样品,比 如掺杂量分别为:0.01%,0.025%,0.05%,0.01%等,分别去做 xrd,如果在 0.05%时出现了杂 质峰,也就是掺杂物质的峰,说明掺杂物质在被掺杂物质中的溶解度在 0.05%左右.11小木虫荣誉出品问题五:XRD拆峰问题如何解决 liuds72: 合成的物质存在多个相,它们每个相的模拟衍射的数据都有了,也得到了他们混合 的物相的 XRD 数据和图谱, 如何将混合的物相的 XRD 图谱拆开, 算出这其中各相的含量. 有这样的 XRD 拆峰软件吗? 回答如下: w6y8d0: 用 Fullprof 做全谱计算,就可以其中各相的含量. Ossolicp: origin 软件也可以.以 origin7.0 为例子,将 peak fitting session 插件托到 origin 界面, 就回出现一个分峰的按钮, 点击, 就可以按照提示将多峰分离, 计算比例. 请楼主试试: ) liuds72: 这样是人为分离不了的,因为多相的晶体粉末数据是很各个独立相的峰形的叠加 的,origin 是处理不了了,因为你根本就不知道那个峰要分开的,不知道那位 DX 知道 search match 软件的用法,好象这个软件有这个功能,恳请帮忙! Jiazang: 其实峰的拆分用 XPS 的分析软件也很好用! 不知 LZ 兄弟是否已经解决了这个问题! LZ 兄弟,我很惭愧,用 XPS 的相关软件拆分 XRD 中相互重叠和屏蔽的峰,只是提供 一种很普通的数学工具,可以将每一个峰的峰位,形状通过拟合的方式提炼出来,可以 确定体系中的物相,这个应该是没有什么问题的.但是如果说要确定每一种物相的相对 含量,这个虽然也有人说 XRD 能够大致测试出来,但是结果可信度很差的,而且鄙人也 没有采用这种方法作过.我可以建议 LZ,在确定物相之后,采用元素分析的方式测出每 一种元素的含量(半定量) ,然后由元素含量来推导每个物相的含量(半定量) .惭愧鄙 人见识浅薄,不知我说的有没有用! Avad: 通过 XPS 分峰,可以确定每个物相的成分,准确程度靠分峰操作控制.如果是同一 个元素形成多个物相,要十分准确区分还是有一定难度,因为不同物相的峰位可能差别 才 1 或者零点几度.要很精确地确定成分,建议使用化学方法.12小木虫荣誉出品XRD 是定性的测试,要计算具体含量,也不是十分现实.通过 Fullprof 是相对成熟 的方法,但是不知道你的单一物相峰是否有重合的? 问题六:关于XRD知识的问题 yanghu5220: 我现在想找一个 JADE 上面没有的物质的 XRD 的标准衍射卡,我知道了该物质的晶 格常数和所属的空间群,可以通过什么软件计算出它的粉末衍射卡? 回答如下: wangzy1000: 一. JADE 上面没有的物质,是说物质是新的还是该物质在 JADE 库中的卡片被删 (JADE 中有些卡片由于各种原因会被删掉但卡片号还存在)或找不到?应该是后者吧. 那你可到别的 XRD 软件如 Search Match 以及 PCPDFWin 等中找啊.如是前者那你的晶 格常数和空间群怎么得到的? 二. 标准谱是用来比对验证的,只有你有你所合成的物质的衍射谱才能比对啊, 那如你已有做谱图,直接比对即可. 三. Xiemax: 楼主是我师兄,他在楼上的高人所说的三个数据库查找了,都没有!但是有一篇文献 说到了他的 xRD,和晶格常数, 但是和另外一篇中又完全不一样! 连所给的 XRD 的都不一 样! !我们也不知道那个是准确的!所以工作都不知道怎么开始!例如:ABO3 和 BAO3 的 XRD 是一样的吗?其中一篇和我们找到的 XRD 一样!(A,B 和 O 都表示元素) wangzy1000: 怎么肯定文献和你的是一样的呢?而且怎么查的?ABO3 和 BAO3 这只是表达的关 系,不和有机物一样与其结构没任何关系,而且同样的分子式可能有几十种甚至几百种 XRD 谱图存在,而且有些谱图差别可能很小.晶格常数等参数须是以自己的 XRD 谱图计 算出来的,而不能参照别人的,结果必须是建立在实验的基础上的.如有扫 XRD 谱,应 先从解谱开始.实在解不出,可以用别的表征手段如 UV-Vis,FTIR,XPS,EDS 等等. 问题七:XRD样品相组成分析 水亦芳: 我做了几个样品,拿去用XRD检测了,出来的数据峰都很明显,但实验员帮我对了标准 如你精于计算的话也可以用晶格常数及群对称性来计算,但那有意义吗?13。
XRD分析软件jade5.0安装
XRD分析软件jade5.0这款XRD分析软件在国内应用是相当多的,估计很多同学或老师刚开始接触XRD的时候,用得最多的XRD分析软件就是MDI JADE5.0了。
MDI JADE5.0的功能非常丰富,一般应用进行的工作,JADE5.0均能胜任。
1、JADE5.0可以进行衍射峰的指标化
2、JADE5.0可以进行晶格参数的计算
3、JADE5.0可以根据标样对晶格参数进行校正
4、JADE5.0能轻松计算峰的面积和质心
5、JADE5.0的出图功能相当强大,你可以在图上进行更加随意的编辑。
XRD分析软件JADE5.0安装“注意事项”:
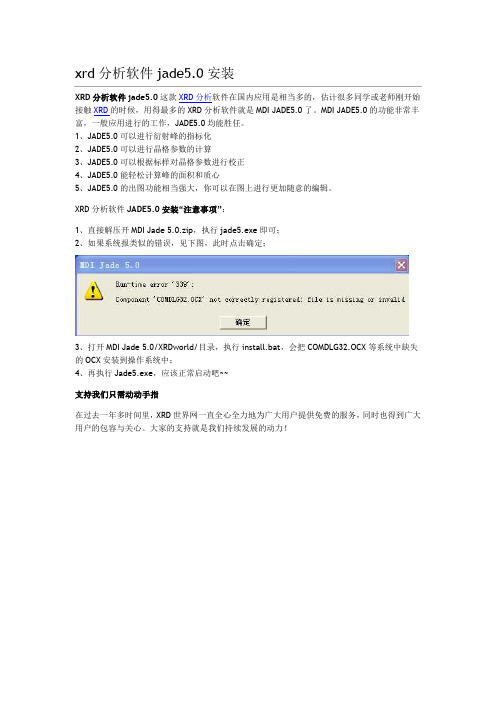
1、直接解压开MDI Jade 5.0.zip,执行jade5.exe即可;
2、如果系统报类似的错误,见下图,此时点击确定;
3、打开MDI Jade 5.0/XRDworld/目录,执行install.bat,会把COMDLG32.OCX等系统中缺失的OCX安装到操作系统中;
4、再执行Jade5.exe,应该正常启动吧~~
支持我们只需动动手指
在过去一年多时间里,XRD世界网一直全心全力地为广大用户提供免费的服务,同时也得到广大用户的包容与关心。
大家的支持就是我们持续发展的动力!。
科研工具-数据分析-XRD分析-FullProf 2020.6安装详细教程

周老师-测试狗科研服务平Next】
(9)点击【Next】
周老师-测试狗科研服务平台技术顾问
• 安装教程
(10)等待一段时间
(11)点击【Finish】
周老师-测试狗科研服务平台技术顾问
• 安装教程
(12)点击【关闭】
(13)软件安装成功,可以开始使用啦
FullProf 2020.6安装教程
此处添加符合主题的图片+logo等
周老师-测试狗科研服务平台技术顾问
软件介绍&获取
FullProf Suite 是 由 FullProf , WinPLOTR , EdPCR , Gfourier 等程序组成的一组晶体学软件 ,适 用 于Windows、 Linux和macOS系统,主要用于X射线粉末衍射精修。
周老师-测试狗科研服务平台技术顾问
• 安装教程
(2)根据自己的系统选择合适的安装包双击运行,本次以64位 win10为例
(3)点击【Next】
周老师-测试狗科研服务平台技术顾问
• 安装教程
(4)点击【Next】
(5)点击【Next】
周老师-测试狗科研服务平台技术顾问
• 安装教程
(6)自定义安装位置,点击【Next】
周老师-测试狗科研服务平台技术顾问
安装教程
安装注意事项
① 安装包说明:此次安装无需断网安装。可自定义安装路径, 但请避免出现中文路径。 ② 安装前请退出所有第三方杀毒软件和安全卫士,安装包无毒 可放心使用。 ③ 配图如有不清晰,请注意参照文字说明 ④ 请打开文件时尽量右键管理员身份运行
(1)下载安装包,右键解压
周老师-测试狗科研服务平台技术顾问
Highscore的使用经验

XRD分析软件Highscore的使用经验2008-09-17 23:21:58| 分类:实验备录|字号订阅X'pert Highscore 是荷兰philips分析仪器公司推出的一款用于xrd物相分析的软件,简便易学。
一.软件安装的时候注意软件和pdf2数据库的连接。
在命令行:customize-programs settings-reference patterns里有两个窗口1. folder with …指得是你的pdf2数据库的位置。
只在进行转换时有用,转换完成后就没有用了。
你可以指定为放置pdf2光碟的光驱。
2. folder for the …指得是转换以后的pdf数据库的位置,系统有个默认的目录,一般不用修改。
进行物相分析时程序从该处调用。
二.进行物相分析的一般套路:1. 打开xrd文件。
Open 菜单里有很多可以打开的文件,highscore默认的文件·rd格式。
2. 峰形处理。
如果峰比较尖锐则只需要把kα2剔除就可以了。
如果峰比较宽化则要慎重处理。
看似一个峰,其实也可能是几个宽化峰叠加。
一般这种峰的轮廓线毛刺很多,要适当的进行平滑等处理。
平滑的标准是既使峰的轮廓线显得清楚,又不使它失真。
尤其是当图谱里既有尖锐的峰,也有宽化的峰的时候,要很小心。
3. 寻峰。
命令行为peak search。
该窗口有一些程序默认的设置,你也可以根据峰的形状设定。
Minumum signficance 最小的意义值。
如果一些肉眼观测到的衍射峰没有寻出来,可以减小此值。
Minumum tip 最小的峰尖宽度。
如果峰较宽化,应当放大该值。
Maximum tip 最大的峰尖宽度。
Peak base 峰背底的宽度,一般为maximum tip的2倍。
如果总有西些个别的峰寻不出来,也可以手动加峰。
命令行为Insert peak如果出现了一些分明不是峰的峰,也可以手动去掉。
在peak list里选中它,然后从键盘上按delete。
XRD分析软件使用手册,使用说明。很好用的哦!

X’Pert HighScore (Plus) 快速入门指南X’Pert HighScoreX’Pert HighScore Plus目录1. 约定术语 (1)1.1. 动作术语 (1)1.2. 指令与说明 (1)1.3. 按钮与表单域 (1)1.4. 菜单项与按键 (1)2. 启动HS+并启用示例数据库 (2)2.1. 介绍 (2)2.2. 启动X’Pert HighScore Plus (2)2.3. 启用数据库 (4)3. 载入及显示数据 (6)3.1. 介绍 (6)3.2. 载入扫描 (6)3.3. 显示扫描 (7)3.4. 检索参考卡片 (8)3.5. 显示参考卡片 (10)4. 图谱处理 (12)4.1. 介绍 (12)4.2. 确定背景 (12)4.3. 寻峰 (14)5. 进行物相鉴定 (17)5.1. 介绍 (17)5.2. 物相检索 (17)5.3. 物相鉴定 (18)6. 改变评分 (21)6.1. 介绍 (21)6.2. 改变评分 (21)7. 运行用户批处理 (22)7.1. 介绍 (22)7.2. 运行用户批处理 (22)8. 检索及精修晶格 (23)8.1. 介绍 (23)8.2. 准备 (23)8.3. 载入图谱 (23)8.4. 寻峰 (25)8.5. 检索/精修晶格 (27)8.6. 保存结果 (30)9. 自动Rietveld精修 (31)9.1. 介绍 (31)9.2. 载入数据 (31)9.3. 自动精修 (32)9.4. 更好的精修 (32)9.5. 注解及建议 (33)10. 物相鉴定策略及疑难解答 (35)10.1. 介绍 (35)10.2. 图谱处理顺序 (36)10.3. 物相鉴定 (37)10.4. 疑难解答 (37)1. 约定术语1.1. 动作术语本指南中代表动作的术语如下:单击按下鼠标左键并马上松开双击快速地重复两次按下鼠标左键并马上松开拖动按下鼠标左键不放,拖动鼠标以划定一个矩形区域或移动一个对象输入打入文本或数字类型的数据按下按下键盘上的一个键或窗口中的一个按钮右击按下鼠标右键并马上松开选中将鼠标指针移到你要的选项或对象上并单击勾选在复选框中打上勾切换来回改变参数或状态(如:开-关-开)在本指南中单击(或按下)意味着关闭你当前工作的窗口而非程序1.2. 指令与说明指令性的文字前面有圆点符“·”。
XRD数据分析 全面详细

5、用Materials studio绘出球棒形 晶体结构
输入晶胞参数:
Sr(SO4)的球棒模型为:
隐藏数据列表:
将Y轴表头改为intensity 将X轴表头改为2theta
二、Search Match检索工具进行物相
点击Search Match 进行分析
进行Байду номын сангаас对比:
进行第二种物相分析
同样进行峰对比
初步确定物相为:
Sr(SO4)ICSD Number: 028055 Na2Zr(Si3O9)(H2O)2 ICSD 现方式做保护处理对用户上传分享的文档内容本身不做任何修改或编辑并不能对任何下载内容负责
《材料现代测试技术》作业-XRD分析 非H、O 成分: Na Zr Si Sr S
作业步骤
1)用orgin绘图工具将图绘出,并正确标出面网间距值(精确到小数点后 四位);
三、从findit软件中找到相应的cif 文件
输入第一种物相的含有的元素Sr、S、O
Sr(SO4)的球棒模型为:
同样Na2Zr(Si3O9)(H2O)2 的球棒模型为:
cif文件的输出:
cif文件为
四、运用maud 进行精修
导入cif文件
4.1 修基线
添加参数至5个
右击参数的value值改成 refined
结果:
4.2修晶胞参数
结果:
4.3修微结构
4.4修原子位子
两种物相每个原子都要改
4.5修择优取向
Sr(SO4) , weight %: 60.28361 +- 0.43405727 Na2Zr(Si3O9)(H2O)2 weight %: 39.71639 +- 0.43405727
MDI-Jade最完整教程(XRD分析)
►PDF 菜单:
▪ PDF-Setup 命令:这个命令的作用是导入ICDD PDF 卡片索引。在拥有一个MDI Jade 软件的同 时,你必须拥有一套ICDD PDF 卡片库。在Jade 作物相检索前,必须导入ICDD PDF卡片索引。这 个命令打开PDF-Setup 对话框,建立PDF 卡片索 引。
► 注意: (1)该命令保存的是窗口中显示的图谱,如果窗口中显示的是
某一个图谱的一部分,那么,保存的只有那么一部分。保存前注意设置
显示为Full Range(View-Zoom Window菜单)。
►
(2)如果保存前作过平滑处理(smooth),则保存的数据为平
滑后的数据而非原始数据。
基本功能操作
峰形拟合:衍射峰一般都可以用一种“钟罩函数”来表示,拟合的意义就是 把测量的衍射曲线表示为一种函数形式。在作“点阵常数精确测量”、“晶 粒尺寸和微观应变测量”和“残余应力测量”等工作前都要经过“扣背 景”——图形拟合”的步骤。常用工具栏中的拟合命令将全谱拟合,但有时 因为窗口中峰太多,计算受阻而不能进行,此时,需要用到手工拟合按钮。 拟合方法在“拟合”一节作祥细介绍。
► File菜单 :
▪ 在这一菜单中,主要命令包括读入数据文件的两种方式Patterns和 Thumbnail。
▪ 另一个特别有用的命令是Save。这个命令具有下级菜单,其中主要的是: ▪ Save-Primary Pattern as *.txt:将当前窗口中显示的图谱数据以文本格
式(*.txt)保存,以方便用其它作图软件如Origin作图和作其它处理。
►因此,通过实验测量或理论计算,建立一个 “已知物相的卡片库”,将所测样品的图谱 与PDF卡片库中的“标准卡片”一一对照, 就能检索出样品中的全部物相。
XRD分析软件使用
XRD分析软件使用XRD(X射线衍射)分析是一种常用的无损表征材料结构的方法,可用于识别晶体结构和晶格参数以及确定晶体中的晶相组成等。
为了进行XRD分析,通常需要使用专门的XRD分析软件。
1.数据质量评估:XRD软件可以对XRD数据进行质量评估,以判断实验数据的准确性和可靠性。
它可以检查峰的形状和位置,并通过计算峰形参数来评估数据的质量。
2.数据处理和曲线拟合:XRD软件可以对原始XRD数据进行处理和分析,包括背景去除、数据平滑和峰位校正等。
此外,软件还可以对XRD曲线进行拟合,以得到准确的晶格参数、晶体结构和相对含量等信息。
3.晶体结构分析:XRD软件可以通过模拟衍射数据进行晶体结构分析。
用户可以输入晶体结构信息,例如原子类型和位置,软件将模拟出理论衍射图案与实验数据进行对比。
通过优化晶格参数和各原子位置,可以得到最佳的拟合结果。
4.相对含量分析:XRD软件可以用于估计样品中各晶相的相对含量。
它可以通过标准样品进行校准,利用衍射峰的强度或面积与样品中晶相的相对含量之间的关系来计算。
5.相图分析:XRD软件可以绘制相图图谱,以直观地展示不同温度、压力和成分条件下的相变和相平衡情况。
这对于材料研究和材料设计具有重要意义。
6.数据库查询:XRD软件通常与各种晶体结构数据库相结合,以便用户能够查询和获得已知晶体结构的相关信息。
这方面的一些知名数据库包括ICSD(国际晶体结构数据库)和PDF(粉末衍射数据库)等。
在使用XRD分析软件时,首先需要导入实验数据,这可以是从X射线衍射仪器中得到的原始数据文件。
然后,进行数据质量评估,包括检查峰的位置和形状等。
接下来,可以对数据进行背景去除和平滑处理,以提高数据的信噪比和可读性。
然后,可以进行曲线拟合,以获得晶格参数、晶体结构和相对含量等信息。
拟合过程通常使用Rietveld方法进行,该方法可以同时考虑衍射强度、衍射峰形和背景等因素。
通过优化晶格参数和各原子位置,可以得到最佳的拟合结果。
XRD分析软件jade5.0应用
相对于其它XRD分析软件,JADE系列软件应该说是做的最好的,它囊括了从定性,指标化,峰型拟合(JADE5.0)到无标定量,晶体结构分析(Jade6.0)等一系列的功能。
我个人以为,只要有Jade5.0就够了,至于后面两项功能,最好是用其它免费软件执行(在上应有尽有),傻瓜相机虽然好用,但要拍出好照片,还是专业相机好。
NO1.Jade5.0的安装和设置Jade5.0都是自动安装的,这不成问题。
要把PDF卡片引入,先将ICDD的光盘插入,然后pdf/setup/select all/,其它按提示进行。
可以对优选项进行设置:EDIT/preference/,里面包括了对显示窗口的设置,仪器参数的设置,打印输出的设置等,一般来说按默认就行,我本人则喜欢将MISC栏里的“Materials Data, Inc.”改为我自己的大名No2 数据的输入Jade软件可以直接读取Rigaku、Bruker、Philips、Scintag等很多衍射仪的原始数据。
打开File\patterns,将出现如附件中所示画面,先(I)找到你文件位置,从(III)的下拉框中选择你的数据格式,按(II)选择。
很多仪器输出文件的格式都是*.raw,实际上都是不一样的,但格式选错了也没关系,软件会给你自动转到合适的格式中去的。
高级一点的:有一些数据格式在(III)的下拉框中没有,比如最常见的txt,xy等,此时你可以自己动手设置,在以上的数据输入面板中,点击工具栏上的“import",进入格式设置画面,如附件所示,a区为注释区,b区为数据格式区,对于最简单的一列角度,一列强度的数据格式,a区不用填写,b区在” angle column“前打上勾,数据从第1行开始读,每行1列数据,强度数据从第8行开始(角度不算),角度从1至6列,所得数据格式即为附件中所示的数据格式。
你也可以按照自己的数据格式进行自由改动,如果a区中表明第1行有说明文字,则数据从第2行读入,相应在b区就将data starts改成2。
XRD物相分析(Jade软件的应用)
第一步:数据的导入、导出处理
选择文件的类型,比
如.txt;.acs文件等,双击所需分析的文
jade软件可打开。
打开后,大家会发现这个是以d值为横坐标的,不是我们常规的2theta角为横坐标。
不能用oringe作图,怎么办?
点开
Primariy
*.txt,就可将原始文件存储为
强度的两列数字保存在txt文件中,导出的文件第一行是文件名称,删除后,即可倒入oringe
第二步:数据的分析
寻峰结果可以帮助后面物相鉴定
最好全选
先确定了主晶相:金红石相TiO
2
确定了第二相:板钛矿相TiO
2
定量分析的计算•采用绝热法进行计算
计算公式有:I
1/I
2
=(R
1
/R
2
)*(w
1
/w
2
)
结晶度计算
结晶度的计算
是不准确的,
这个数字可以
直接获得结晶
度的大小信
息,但是受到
背底扣除的极
大影响。
平均晶粒大小计算
注意,计算时需要对不重叠的峰进行
计算。
如果都是重叠的,则需要进行
分峰再进行计算。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
XRD分析软件有4种
1.pcpdgwin 有人认为是最原始的了。
它是在衍射图谱标定以后按照d值检索。
一般可以有限定元素、按照三强线、结合法等方法。
所检索出的卡片多时候不对。
一张复杂的衍射谱有时候一天也搞不定。
2.search match 可以实现和原始实验数据的直接对接可以自动或手动标定衍射峰的位置对于一般的图都能很好的应付。
而且有几个小工具使用很方便。
如放大功能、十字定位线、坐标指示按钮、网格线条等。
最重要的是它有自动检索功能。
可以帮你很方便的检索出你要找的物相。
也可以进行各种限定以缩小检索范围。
如果你对于你的材料较为熟悉的话对于一张含有45相的图谱检索也就3分钟。
效率很高。
而且它还有自动生成实验报告的功能
3.High Score 几乎search match中所有的功能highscore都具备而且它比searchmatch更实用。
1它可以调用的数据格式更多。
2窗口设置更人性化用户可以自己选择。
3谱线位置的显示方式可以让你更直接地看到检索的情况 4手动加峰或减峰更加方便。
5可以对衍射图进行平滑等操作是图更漂亮。
6可以更改原始数据的步长、起始角度等参数。
7可以进行0点的校正。
8可以对峰的外形进行校正。
9可以进行半定量分析。
10物相检索更加方便检索方式更多。
11可以编写批处理命令对于同一系列的衍射图一键搞定。
4.jade 和highscore相比自动检索功能少差但它有比之更多的功能。
1它可以进行衍射峰的指标化。
2进行晶格参数的计算。
3根据标样对晶格参数进行校正。
4轻松计算峰的面积、质心。
5出图更加方便你可以在图上进行更加随意的编辑。
其实学化工和材料的人对这个软件HighScore是很熟悉的还有一个软件数据分析与科学绘图软件origin 是搞材料研究算是必备的软件。
对于学材料学生可能毕业写论文要用到这些软件。
Jade5.0的使用初步说明 1、数据输入由于不同的X射线衍射仪输出的数据类型不同但都可以将数据转换成txt文档或Ascii格式的文档文件名为.txt 或.asc为提高软件的通用性jade5.0提供了以txt文档或Ascii格式输入数据。
运行jade5.exe首先进入以下界面图片一在后面的压缩文件里面下同中间的窗口用于选择需打开文件左侧选择文件路径与资源管理器的操作相同右侧选择打开文件的类型一般选择XRD Pattern files.这时在右下方的窗口中将显示左侧被选择文件夹中所有能被该软件识别的文件然后选择需要分析的数据文件点击菜单栏Read进入主窗口此选择窗口可以通过主窗口中file/patterns进入。
2、背景及Ka2线扣除在主菜单栏中选择analyze/fit background进入如下窗口图片二该窗口用于设置扣除背景时的参数一般选择默认值直接选择apply回到主窗口此时软件自动运行Edit bar/B.E按钮用于手动修改背景 Edit bar工具栏如下图片三此工具栏提供了放大、标定峰位等操作当鼠标移动到按钮上时软件将自动提示。
在该软件中的所有按钮对鼠标左右键操作都有不同效果
一般左键为确定或正向操作右键为取消或反向操作。
3、确定峰位在主菜单栏中选择analysie/find peaks进入确定峰位所需的参数设置窗口如下图一般选择默认值选择apply回到主窗口选择Edit bar左第三个按钮可手动编辑。
图片四在手动编辑过峰个数或峰位后同样可以选择analyze/find peaks 选择Report进入如下界面图片五在此窗口中显示了以上操作中所确定的峰位置、强度、半峰宽FWHM等参数其中FWHM将时计算晶粒度的主要参数。
选择analyze/find peaks在此窗口中选择Labeling标签可以选择峰的标示方式如下图图片六通过以上操作主窗口将为如下效果图片七 4、PDF数据库加载与定性分析做定性及定量分析之前需要将PDF数据库载入软件在主窗口中选择PDF/Setup将显示如下窗口图片八在其中选择PDF数据库所存储的位置及所需加载PDF卡片的种类。
载如PDF数据库后选择主菜单Identify/Search Match setup进行测试数据与标准图库匹配过程将显示Search /Match Display窗口在其中选择相匹配的相。
6、定量分析在主菜单中选择analyze/Theta Calibration在出现的窗口中选择标定数据的方式在Options中选择要进行的定量分析如晶面指数、晶粒度、晶胞参数等方面的计算其具体步骤可参阅软件自带的帮助文件jade5.chm。
关于jade5.0数据修改说明每种设备输出的都有其特殊的格式但都可以输出以.txt格式的数据文件下面说明一下把.txt转换成jade通常识别的.mdi文件的方法 1 、一般.txt开始是说明测试数据保存两列一列按步长的测试点一列测试结果用文本文档的方式打开jade中提供的.mdi文件会发现只有一列测试的结果因此需要把.txt中的测试结果拷出来 2、因为在.txt文件中不能整列拷数据以下说明把.txt中的测试结果拷出来的方法首先把.txt文件后缀改成.xls这样就用excle打开文件把前面的测试说明删除只保留数据这时两列数据在一列表格中按以下步骤选择该列数据分列分割符然后根据不同的选择分割符合适的分割符有时候是逗号有时候是空格 3、把测试点复制到一个.txt文件中第一行空下接下来用文本文档打开.mdi文件按照其格式及数据测试填写第二行经过修改的.txt文档格式为空行起始角步长 1.0 衍射靶 x射线波长终止角测试点个数测试结果 4、保存修改好的.txt文档把后缀txt改成mdi即可直接使用jade打开。
Highscore的使用经验 Xpert Highscore 是荷兰philips分析仪器公司推出的一款用于xrd物相分析的软件简便易学。
是我见到的物相检索最好的软件了。
现在给大家讲一点我的使用经验希望能对您有些许帮助同时求证于方家有不对的地方敬请指正。
一软件安装的时候注意软件和pdf2数据库的连接。
在命令行customizeprograms settingsreference patterns里有两个窗口
1 . folder with …指得是你的pdf2数据库的位置。
只在进行转换时有用转换完成后就没有用了。
你可以指定为放置pdf2光碟的光驱。
2. folder for the …指得是转换以后的pdf数据库的位置系统有个默认的目录一般不用修改。
进行物相分析时程序从该处调用。
二进行物相分析的一般套路 1. 打开xrd文件。
Open 菜单里有很多可以打开的文件highscore默认的文件·rd格式。
2. 峰形处理。
如果峰比较尖锐则只需要把kα2剔除就可以了。
如果峰比较宽化则要慎重处理。
看似一个峰其实也可能是几个宽化峰叠加。
一般这种峰的轮
廓线毛刺很多要适当的进行平滑等处理。
平滑的标准是既使峰的轮廓线显得清楚又不使它失真。
尤其是当图谱里既有尖锐的峰也有宽化的峰的时候要很小心。
3 . 寻峰。
命令行为peak search。
该窗口有一些程序默认的设置你也可以根据峰的形状设定。
Minumum signficance 最小的意义值。
如果一些肉眼观测到的衍射峰没有寻出来可以减小此值。
Minumum tip 最小的峰尖宽度。
如果峰较宽化应当放大该值。
Maximum tip 最大的峰尖宽度。
Peak base 峰背底的宽度一般为maximum tip的2倍。
如果总有西些个别的峰寻不出来也可以手动加峰。
命令行为Insert peak 如果出现了一些分明不是峰的峰也可以手动去掉。
在peak list里选中它然后从键盘上按delete。
4物相检索。
命令行为Search and match 其中有很多设置。
1 restrictions里 none是没有任何限定。
Restriction set是元素限定点击edit 按钮会弹出相应的对话框。
Subset 是限定设置文件需要事先编辑。
其他的按钮等你熟悉了再改动它。
2parameters里 data source一般选用peak and profile date 如果你试样是单相选择single phase 否则选择 multi phase 其他的暂时不动等熟悉了再说。
3retrieve patterns by text search 名称查询。
比如你要查询石英的卡片可以在这个对话框里输入quartz并选mineral name。
4retrieve patterns by reference code 是直接输入卡片号。
看你的衍射谱里有没有该卡是标定的物质。
5auto residue按钮是很重要的。
选中它则自动寻找剩余峰。
如你的试样里有a相和b相。
当你选择了a相后程序会自动在衍射谱里寻找a相以外的相。
你可以从它列出的选项里很容易的找出b相。
5. 文件的保存。
Highscore程序默认的格式是caf格式只有这种格式可以保存你检索的全部信息。
其他的格式只是能保存你处理过的峰的形状而已。