转录组学分析流程及常用软件介绍.pdf
转录组分析流程范文

转录组分析流程范文转录组分析是一种用于研究特定生物体或组织中的全部转录本的方法。
它通过测量RNA分子在特定时间点和条件下的表达水平,揭示了基因转录的调控机制,功能注释和转录本的表达图谱,并为找到新的基因和功能注释提供了线索。
下面是一个常用的转录组分析流程,包含预处理、质控、序列比对、表达定量和差异分析等步骤。
1.数据预处理:数据预处理主要包括质量控制和去除低质量序列。
在高通量测序中,原始的测序数据通常包含低质量碱基、接头序列和多聚A/T等噪声。
质控工具如FastQC可以用于评估测序数据的质量,根据其结果,采用切割、修剪、过滤和去除低质量序列来提高数据质量。
2.序列比对:序列比对是将预处理后的测序数据与参考基因组进行比对的过程。
常用的比对工具包括Bowtie、BWA和STAR等。
这一步的目的是将测序数据映射到参考基因组上,以确定每个读取序列的起始位置。
3.表达定量:表达定量是将比对得到的序列转化为表达量的过程,它衡量了每个转录本的相对丰度。
常用的表达定量工具包括HTSeq、Cufflinks和Salmon 等。
这些工具通过计算每个转录本对应的读取数来估计其表达水平,并生成基因表达矩阵。
4.差异表达分析:差异表达分析是比较不同条件下的基因表达量,以确定哪些基因在不同条件下的表达水平发生了显著变化。
常用的差异表达分析工具包括DESeq2、edgeR和limma等。
这些工具通过统计学方法来鉴定不同条件之间的显著差异,并生成差异表达基因列表。
5.功能注释:功能注释是对差异表达基因进行生物学功能注释的过程。
常用的功能注释工具包括DAVID、GOseq和KEGG等。
这些工具通过富集分析、与已知数据库的比对和基因网络分析等方法,帮助研究人员了解差异表达基因的功能和参与的生物学过程。
6.结果可视化:结果可视化是将转录组分析结果以图形化的形式呈现,以帮助研究人员理解和解释数据。
常见的结果可视化工具包括Heatmap、Venn图、火山图和基因网络图等。
RNA-seq(转录组学)的分析流程和原理

RNA-seq(转录组学)的分析流程和原理在开始详细讲解RNA测序之前,我们先来了解一下它的基本步骤:1.建库:提取RNA,富集mRNA或消除rRNA,合成cDNA和构建测序文库。
2.测序:然后在高通量平台(通常是Illumina)上进行测序(每个样本测序reads在DNA测序中,读数是对应于单个DNA片段的全部或部分的碱基对(或碱基对概率)的推断序列。
深度为10-30 Million reads。
)3.分析:先比对/拼装测序片段到转录本,通过计数、定量,样本间过滤和标准化,以进行样本组间基因/转录本统计差异分析。
大致了解这个过程之后,我们就先从建库开始了解建库的难点在于提纯出mRNA, 一般在我们抽离出的RNA中rRNA占比很大,其他还会有tRNA、microRNA等。
我们需要从抽离出的RNA中提取出mRNA,并建立cDNA文库。
这里以应用最广泛的Illumina公司的Truseq RNA的建库方法为例来进行介绍。
首先,利用高等生物的mRNA通常有poly(A)尾的(使mRNA更稳定,翻译不容易出错)特点,用带有poly(T)探针的磁珠与总RNA进行杂交,这样磁珠就和带poly(A)尾巴的mRNA结合在一起了。
接下来,就回收磁珠,把这些带poly(A)的mRNA从磁珠上洗脱下来。
再用镁离子溶液(或者超声波)进行处理,把mRNA打成小段。
然后,利用这些被打断的mRNA片段,以随机引物进行逆转录,得到第一链cDNA。
再根据第一链cDNA合成出ds-cDNA。
对cDNA在平末端进行3’端加A碱基(腺苷酸)(adapter接头上带了T碱基头,为了和adapter配对)在双链cDNA的两端加分别上Y型接头再经PCR扩增经筛选的目的基因,就得到可以上机的测序文库了。
这个建库方法对RNA的完整度有较高的要求。
也就是说,只有在mRNA大部分是完整的状态下,才能得到比较好的效果。
因为带Poly(T)的磁珠,它所吸附的是带有Poly(A)的那些序列。
转录组测序数据分析及其应用

转录组测序数据分析及其应用基因组学研究一直是生命科学领域的重要分支,而随着高通量测序技术的发展,转录组学研究也变得越来越重要。
转录组测序是一种高通量的测序技术,可用于分析RNA的产生和使用。
它可以用于解析基因表达调控机制、鉴定新的转录产物、发现新的基因及其功能以及研究基因表达的变化。
而分析和处理转录组测序数据则是实现这些研究目标的关键步骤。
本文将介绍转录组测序数据分析的流程以及其在生命科学研究中的应用。
1. 转录组测序数据分析的流程转录组测序数据分析的流程包括测序数据质量控制、选用合适的参考基因组进行比对、基因表达量计算、差异表达基因筛选、功能注释及通路分析等步骤。
(1)数据质量控制在数据分析之前,需要对原始测序数据进行质量控制。
质量控制通常包括检查样品的测序深度和比对率,检测是否存在序列重复和序列污染,并通过统计和图形化分析来评估测序数据的准确性和一致性。
一些常用的工具如FastQC和Trimmomatic可以用于数据质量控制。
(2)比对与注释转录组测序数据的比对和注释是数据分析过程中的关键步骤,它可以帮助我们理解基因组中那些区域正在表达这些转录物,并且可以使下游分析过程更加准确和可靠。
常用的比对软件有TopHat和STAR等,同时,基于火山图和MA-plot等绘图技术,对比对结果进行筛选与统计分析,即可确定差异表达的基因。
(3)差异表达基因筛选通过比对和注释分析后,我们可以通过基因表达量的计算来确定哪些基因在不同的实验条件下差异表达。
常用的对基因表达量计算的方法有FPKM和TPM等,同时也适用于多样品比较的统计方法如edgeR和DESeq2等,以筛选差异表达的基因。
(4)功能注释及通路分析结合差异表达基因的结果进行进一步的功能注释和通路分析,通过各种生物信息学工具对其进行KEGG、GO、Cytoscape等分析,以便确定关键的基因、分子和通路在生物学过程中的作用。
2. 转录组测序数据的应用转录组测序数据被广泛应用于生命科学领域中的多种研究,如基因组结构与表达、药物研发、癌症研究、农业作物育种、蛋白质组学、环境科学等等。
转录组分析报告

转录组分析报告介绍转录组分析是研究基因组中转录过程的研究领域。
通过转录组分析,我们可以了解到在特定条件下细胞中正在转录的所有基因。
这些信息对于理解细胞功能、疾病发展以及生物技术的开发都非常重要。
本报告将介绍转录组分析的一般步骤和常用方法。
步骤一:实验设计转录组分析的第一步是设计实验。
在这个步骤中,我们需要确定要研究的样本类型、实验条件和重复次数。
合理的实验设计可以最大程度地减少误差,并提高结果的可靠性。
步骤二:RNA提取在转录组分析中,我们需要从样本中提取RNA。
RNA是细胞中转录的产物,它可以反映细胞中正在表达的基因信息。
RNA提取的质量和纯度对后续的转录组分析非常重要。
常用的提取方法包括酚氯仿法、磁珠法和硅胶膜法等。
步骤三:RNA测序RNA测序是转录组分析的核心步骤之一。
通过RNA测序,我们可以将RNA样本转化为对应的DNA序列,并确定每个基因的表达水平。
常见的RNA测序技术包括Sanger测序、二代测序和三代测序等。
二代测序技术如Illumina和Ion Torrent等已经成为转录组分析的主流技术。
步骤四:数据预处理RNA测序会产生大量的原始数据,这些数据需要进行预处理以去除噪音和提高数据质量。
数据预处理包括去除低质量的reads、去除接头序列、去除重复序列和过滤低表达基因等。
预处理后的数据可以为后续的分析提供可靠的基础。
步骤五:差异表达基因分析差异表达基因分析是转录组分析的重要环节之一。
通过比较不同条件下基因的表达水平,我们可以找到与特定条件相关的差异表达基因。
常用的差异表达基因分析方法包括DESeq、edgeR和limma等。
这些方法可以帮助我们发现与特定条件相关的生物学过程和信号通路。
步骤六:功能注释和富集分析一旦确定了差异表达基因,我们可以对这些基因进行功能注释和富集分析。
功能注释可以帮助我们了解差异表达基因的功能和参与的生物学过程。
而富集分析可以帮助我们发现差异表达基因在特定功能和通路中的富集情况。
转录组学分析流程及常用软件介绍

转录组学分析流程及常用软件介绍转录组学是研究在特定条件下生物体内转录的所有RNA分子的总体,包括信使RNA(mRNA)、转运RNA(tRNA)、核糖体RNA(rRNA)和小核RNA(snRNA)等。
转录组学研究可以通过分析转录组中的基因表达水平和调控机制,揭示基因功能和调控网络,从而深入了解生物体的生命活动和适应能力。
转录组学分析流程包括实验设计、RNA提取、RNA测序、数据分析和结果解释等环节,并依赖于一系列的软件工具来完成。
下面将介绍转录组学分析的流程以及常用的软件。
1.实验设计:确定研究目的和假设,设计实验方案,包括样本的选择和处理方式等。
2.RNA提取:从样本中提取总RNA,并进行纯化和富集,去除DNA和其他杂质。
3. RNA测序:将提取得到的RNA反转录成cDNA,然后通过高通量测序技术进行测序。
常用的测序技术包括Illumina HiSeq、Ion Torrent Proton等。
4.数据分析:对测序得到的数据进行质控、比对和定量等处理。
这一步通常需要使用一系列的转录组学分析软件。
5.结果解释:根据数据分析的结果,进行差异表达基因的筛选、基因富集分析和信号通路分析,以探索转录组的生物学意义。
常用的转录组学分析软件包括:1. 基因表达微阵列分析:在早期的转录组学研究中,基因表达微阵列是常用的分析方法。
常用的分析软件有Affymetrix Expression Console、Partek Genomics Suite等。
2. RNA测序数据分析:随着高通量测序技术的发展,RNA测序已成为转录组学研究的主要方法。
RNA测序数据的分析可以分为质控、比对和定量等环节。
常用的软件工具有Trimmomatic、FastQC、STAR、HISAT等。
3. 差异表达基因分析:差异表达基因是通过比较不同样本之间的基因表达水平而筛选出来的。
常用的软件包括DESeq2、edgeR、limma等。
4. 基因富集分析:基因富集分析可以帮助我们了解不同基因集之间的功能和通路差异,从而揭示转录组的生物学意义。
转录组测序数据分析流程
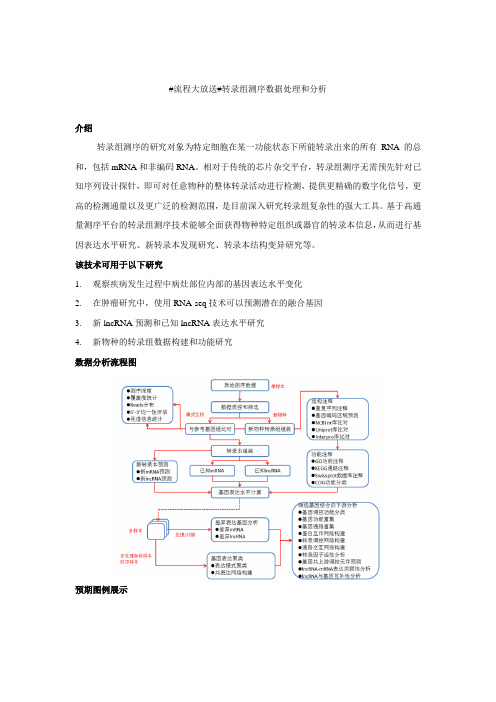
#流程大放送#转录组测序数据处理和分析
介绍
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。
基于高通量测序平台的转录组测序技术能够全面获得物种特定组织或器官的转录本信息,从而进行基因表达水平研究、新转录本发现研究、转录本结构变异研究等。
该技术可用于以下研究
1.观察疾病发生过程中病灶部位内部的基因表达水平变化
2.在肿瘤研究中,使用RNA-seq技术可以预测潜在的融合基因
3.新lncRNA预测和已知lncRNA表达水平研究
4.新物种的转录组数据构建和功能研究
数据分析流程图
预期图例展示
示例图1 差异表达基因筛选示例2 基因聚类分析heatmap图
示例3 差异基因互作网络图示例4 lncRNA、基因与上游共有miRNA网络图。
转录组学分析流程及常用软件介绍

--bowtie-path #bowtie2路径
RSEM用法
• 3.[rsem-calculate-expression]
rsem-calculate-expression [options] upstream_read_file(s) reference_name sample_name rsem-calculate-expression [options] --paired-end upstream_read_file(s) downstream_read_file(s) reference_name sample_name
比对——Tophat2 Tophat2
特点如下: -不依赖于splice sites信息 可只使用参考基因组进行比对; 可以处理spliced reads; -可以利用注释信息进行比对 -序列比对;Bowtie2,也可以使用Bowite -处理错误比对,eg:假基因 -处理Indels
比对——Tophat2
--phred33-quals 、--phred64-quals、 --paired-end
RSEM用法
• extract-transcript-to-gene-map-from-trinity trinity_out_dir.Trinity.fasta isoform2gene • rsem-prepare-reference --bowtie2 --bowtie2-path /PUBLIC/software/RNA/bowtie2-2.2.3 --transcript-to-gene-map isoform2gene --no-polyA trinity_out_dir.Trinity.fasta Trinity.fasta • rsem-calculate-expression --bowtie2 --bowtie2-path /PUBLIC/software/RNA/bowtie2-2.2.3/ --phred33-quals -p 8 --forwardprob 0.0 --time --paired-end reads.left.fq reads.right.fq Trinity.fasta Sample
转录组分析流程范文

转录组分析流程范文1.实验设计:首先,确定要研究的生理条件或比较的样本,例如不同时间点、不同组织、不同处理等。
根据实验设计制定方案,明确实验的目的和假设。
2.样本采集与RNA提取:根据实验设计获取生物样本,例如组织切片、细胞培养等。
通过RNA提取技术将样本中的总RNA提取出来,并进行纯化处理,去除DNA和其他外源性RNA。
3. 文库构建与测序:将提取得到的RNA进行反转录和合成cDNA,然后引入测序适配体,构建转录组文库。
常用的测序技术包括RNA-seq、SAGE、CAGE等。
其中,RNA-seq技术能够对整个转录组进行测序,因此广泛应用于转录组分析。
4. 数据预处理:测序得到的原始数据需要进行质量控制和去除低质量读段、去除接头序列等预处理步骤。
可使用FastQC等软件进行质量评估,然后使用Trimmomatic、Cutadapt等工具进行修剪和过滤。
5. 游离3'末端修正:mRNA分子具有3'末端的短多聚A尾,而RNA-seq测序往往会出现3'末端错误末尾,即PolyA尾的长度不一致。
因此需要进行3'末端修正,并截断导致的测序误差。
6. 比对与定量:将修正后的测序reads与参考基因组或转录组序列进行比对。
比对软件有Bowtie、Tophat、HISAT等。
比对后,可以通过拟合对reads进行定量,比如算法RSEM、Cufflinks等。
7. 差异表达基因分析:通过比较不同条件下的表达量来确定差异表达基因。
一般采用DESeq2、edgeR、limma等软件来进行统计学分析。
8.功能注释与富集分析:对差异表达基因进行生物学功能注释,如基因本体论(GO)、通路富集分析(KEGG)等,找出富集的GO和KEGG通路,以及参与其功能的关键基因。
9.可变剪切分析:转录组测序可以对基因的可变剪切进行研究,通过比对和拼接转录本,找出不同剪切形式的外显子、内含子和连接顺序变化等信息。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• --SS_lib_type (Strand-specific libFra bibliotekary type)
– Paired reads: • RF: first read(/1) of fragment pair is sequenced as antisense(reverse(R)), and second read(/2) is in the sense strand(forward(F)); typical of sequencing method. • FR: (reverse)
len: length of the transcript sequence
Trinity拼接质量评估
• N50/N90:按照长度将拼接转录本从大到小排序,累加转录本的 长度,到不小于总长50%/90%的拼接转录本的长度就是N50/N90。
OUTLINE
拼 接——Trinity(无参) 比对定量——RSEM(无参) 比对软件(Tophat2有参) ) 定量(HTSeq有参)
常用数据库介绍(NCBI,ENSEMBL)
step1:Inchworm
1.分解测序reads,构建k-mer字典 2.从k-mer字典中移除error-containing k-mer 3.选择seed k-mer 4.Seed k-mer 延伸,构成contig 5. 重复seed selection 和 bidirectional k-mer extension 直到k-mer 字 典耗尽 6. 过滤 contig
– Unpaired (single) reads: • F: the single read is in the sense (forward) orientation • R: the single read is in the antisense (reverse) orientation
step2:Chrysalis
1.将contigs 组合成connected components 2. 将每个component构成一个de Bruijn graph 3.reads回比 4.过滤
step3:Butterfly
Butterfly resolves alternatively spliced and paralogous transcripts
--min_kmer_cov 2 \ --min_glue 2 \ --full_cleanup \#删除中间文件
Trinity示例
• 输出结果
– Trinity.fasta文件 – unigene.fasta文件
从trinity.fasta中选择最长的转录本作为unigene,Trinity作者推荐
Trinity参数
• --min_contig_length default:200 (step1,filter contigs)
– minimum assebled contig length to report
• --min_glue default:2 (step1,contigs to componets)
常用数据库介绍(NCBI,ENSEMBL)
1. 图形化简
2.解图,确定转录本序列
Butterfly
•
2016-1-11
Butterfly
2016-1-11
Trinity
• --jaccard_clip
Trinity参数
– for gene-dense compact genome, such as fungal genomes, where transcripts may often overlap in UTR regions.
Trinity拼接结果解读
• >c1_g1_i1 len=233 path=[94:0-232]
c1: sequence is derived from Chrysalis component 1 g1: sequence also corresponds to Butterfly subcomponent# 1 (during graph compaction and pruning, some components are partitioned into disconnected subcomponents). i1: sequence count from chrysalis component 1, butterfly subcomponent 1. If this subcomponent yields multiple sequences, these will have different seq numbers.
转录组分析流程及常用软件使用方法
(无参,有参)
Novogene 孙福明
2015.1.12
无参转录组分析流程
有参分析流程
OUTLINE
拼 接(无参) 比对定量(RESM无参) 比对软件(Tophat2有参) ) 定量(HTSeq有参)
常用数据库介绍(NCBI,ENSEMBL)
OUTLINE
拼 接——Trinity(无参) 比对定量(RESM无参) 比对软件(Tophat2有参) ) 定量(HTSeq有参)
- min numbler of read needed to glue two inchworm contigs together
Trinity示例
ln -s /BJPROJ/RNA/rna_test/TR_bioinfomatics1/prepare/sunfuming/lession5/trinitydata/re ads.*.fq . perl /PUBLIC/software/public/Assembly/trinityrnaseq_r20140413p1/Trinity \ --seqType fq \ --JM 2G \ --left reads.left.fq \ --right reads.right.fq\ --SS_lib_type RF \ --CPU 4 \