金属有机化学第七章
有机化学第四版陈宏博答案第七章

有机化学第四版陈宏博答案第七章简介本文档是有机化学第四版陈宏博答案第七章的概要和重点解析。
第七章主要介绍了有机物分子间相互作用的情况,包括氢键、范德华力、离子键和共价键等。
氢键氢键是有机化学中一种重要的分子间作用力。
它是通过氢原子和一对电负性原子(如氮、氧或氟)之间的相互作用而形成的。
氢键的形成需要满足一定的条件:氢原子与电负性原子距离适中,形成一条直线或接近一条直线;氢键的角度一般是线性的;氢键的能量较高,通常在5-30 kJ/mol范围内。
氢键可以影响分子的性质和反应性。
它可以使分子的凝聚能力增强,从而影响物质的物理性质,如沸点、溶解度等。
氢键还可以作为反应中的重要中间体,影响反应速率和选择性。
范德华力范德华力是一种分子间相互作用力,主要由分子之间的瞬时极化引起。
它是一种弱的相互作用力,通常远小于氢键的能量。
范德华力可以分为两种类型:极化-极化相互作用和极化-非极化相互作用。
极化-极化相互作用是两个极性分子之间的相互作用,极化-非极化相互作用是极性分子与非极性分子之间的相互作用。
虽然范德华力很弱,但它可以累积,并且在分子间相互作用中起到重要的作用。
范德华力可以影响分子的相对位置和取向,从而影响化学反应的速率和选择性。
离子键离子键是一种通过正负电荷之间的相互作用而形成的分子间相互作用力。
它是由正离子和负离子之间的静电相互吸引力所引起的。
离子键具有很强的相互作用力,通常能量超过100 kJ/mol。
它的特点是具有高熔点和沸点,且在溶液中能够电离。
离子键可以影响物质的物理性质和化学反应。
具有离子键的化合物一般具有良好的溶解性,并且在水中能够电离,形成电解质溶液。
共价键共价键是最常见的一种分子间相互作用力,是通过共用电子对而形成的。
它是最强的一种相互作用力,能量通常高于200 kJ/mol。
共价键的形成需要满足规则的电子配对原则和静电的吸引力。
共价键可以稳定分子的结构,并影响分子的大小和形状。
共价键对于有机化学反应至关重要。
有机化学-卤代烃

(4)被硝酸根取代
RX + AAgg+NOONO32— 醇
R ONO2 + AgX
硝酸酯
•卤化银沉淀产生,反应可作为卤代烃的鉴别反应。
卤代烃反应活性: 烯丙基卤>叔卤代烃>仲~>伯~
(5)卤素交换
丙酮
R-Br(Cl) + KI
R-I + NaBr(Cl)
难溶于丙酮
难溶于丙酮
(6)被氨基(-NH2)取代
三、命名
普通命名法 俗名 系统命名法
1.普通命名法
在烃基名称之前(或后)加上卤素的名 称,称为卤(代)某烃或某烃基卤。
CHCl3
三氯甲烷 氯 仿 (俗名)
C2H5Cl
氯乙烷 乙基氯
(CH3)3CBr
叔丁基溴
CH2=CHBr 溴乙烯
H2C CH CH2 烯丙基溴
Br
Br
溴苯
CH3
Br
邻-溴甲苯(2-溴甲苯) 2-bromotoluene
3-甲基-5-氯庚烷 3-chloro-5-methylheptane
H2C C CH3
Cl
2-氯丙烯 2-chloro-propene
CH3CH CHCH2Cl
1-氯-2-丁烯 1-chloro-2-butene
H37C
CH
6
CH
5
C4H2
CH
3
CH
2
C1H3
CH3 I
Cl CH3
2,6-二甲基-3-氯-5-碘庚烷
•(一) 亲核试剂:具有孤对电子的物质, 能与底物中带部分正电荷的碳发生反应, 这 种 物 质 称 为 亲 核 试 剂 。 ( 也 是 Lewis 碱)。
有机化学——第7章醇酚醚

25
6、氧化脱氢反应
在有机化合物的分子中加入氧或脱去氢的反应都叫做氧化反应。
1) 伯醇氧化生成醛,醛进一步氧化生成酸。
CH3 CH2 CH2OH
K2CrO7-H2SO4
CH3 CH2CHO
[O]
CH3 CH2 COOH
2) 仲醇氧化生成酮,酮不易被继续氧化 。
H3 C CH OH CH3 [O] CH3 C O CH3
烯丙位 苯甲位 一级醇
}
醛
弱碱,反应条件温和, 不饱和键不受影响。
琼斯试剂 CrO3+稀H2SO4
费慈纳-莫法特试剂
醛(产率不高, 不用。)
醛(产率很高)
稀酸,反应条件温和, 不饱和键不受影响。 酸性 (H3PO4) , 其它基团不受影响。 碱性,可逆,分子内 双键不受影响。 28
反应机理 1oROH (SN2)
SN2
SN2
BrCH2CH3 +
2oROH , 3oROH (SN1)
SN2
SN1
(CH3)3C+ + HOPBr2
Br -
(CH3)3CBr
18
ROH + SOCl2
b.p. 79oC
RCl + SO2 + HCl
该反应的特点是:反应条件温 和,反应速率快,产率高,没 有副产物。
13
2、与氢卤酸反应
发生亲核取代反应,生成卤代烃和水,是制取卤代烃的重要方法。
R OH + HX
R X + H2O
氢卤酸的活性次序:HI > HBr > HCl; 醇的活性次序:烯丙式醇,苄基醇 > 3º 醇 > 2º 醇 > 1º 醇。 烯丙醇、叔醇、大多数仲醇及空间位阻大的伯醇,反应是按SN1
有机化学课后习题答案7第七章答案
4.
V2O5, O2
一. 命名或写出结构式
1.
2. C2H5
NO2
Br2 Fe
NO2 Br
O
O
O
O
浓H2SO4
AlCl3 O
COOH
O
习题 B 答案
CH3 3.
OH 4.
SO3H
H3C
5.
6.
7. 2-乙基-9,10-蒽醌 8. 2-环丙基萘
9. 1,4-二甲基萘 10. 邻苯二甲酸酐
二.用休克尔规则判断下列化合物是否有芳香性
CHO
CHO
CH3 NBS
O2, V2O5 400-500℃
CH2MgBr 无水乙醚
CH2Br Mg 无水乙醚
CH2MgBr
O
O AlCl3
O
O Zn-Hg HCl
HOOC
H2SO4 HOOC
H3O+
H2/Ni HO CH2
H2SO4 HO CH2
O CH2
5.
O
O
Zn-Hg
浓H2SO4
O AlCl3
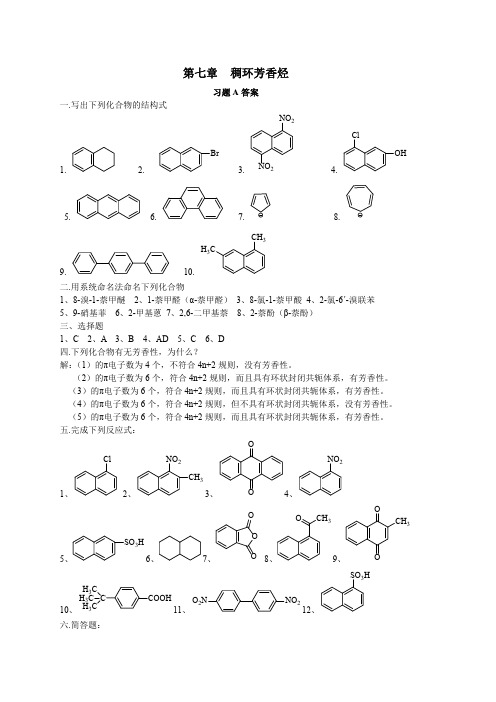
第七章 稠环芳香烃
一.写出下列化合物的结构式
习题 A 答案
NO2
Cl
Br
OH
1.
2.
3. NO2
4.
5.
6.
7.
8.
CH3 H3C
9.
10.
二.用系统命名法命名下列化合物 1、8-溴-1-萘甲醚 2、1-萘甲醛(α-萘甲醛) 3、8-氯-1-萘甲酸 4、2-氯-6ˊ-溴联苯 5、9-硝基菲 6、2-甲基蒽 7、2,6-二甲基萘 8、2-萘酚(β-萘酚) 三、选择题 1、C 2、A 3、B 4、AD 5、C 6、D 四.下列化合物有无芳香性,为什么? 解:(1)的π电子数为 4 个,不符合 4n+2 规则,没有芳香性。
金属有机配合物

7-1 金属茂及其催化不对称合成
环戊二烯的负离子, 即 环戊二烯基叫茂, 记作 Cp-。 20 世纪初 ,Wilkinson 等人发现 Cp- 和 Fe2+ 反应生成 Cp2Fe, 叫铁 茂。
这类金属环戊二烯基化合物, 统称为金属茂。
金属茂化学蓬勃发展起来,开创了近代金属有机 化学的新时代。因此 ,Wilkinson 获得了 1973 年的诺贝尔化学奖。
第七章 金属有机配合物
定义:至少含有一个金属-碳键的化合 物称为金属有机配合物
范畴:无机化学和有机化学的交叉学科
金属有机化学研究的主要内容: 过渡金属和稀土元素配合物
金属有机化合物合成和结构多种多样;促进 了基础化学的发展
在工业、精细有机合成、催化剂、新型功 能材料的开发、生命科学等方面具有重要 意义。
➢ 通过围绕键的旋转而产生的分子中原子或基团在 空间的不同排列方式,称为构象。
➢ 其中较稳定的结构,称为该化合物的构象异构体。 ➢ 上下两个茂旋转,形成一系列构象,相对夹角为构象
角α ➢ α=0°为覆盖型 ➢ α =36°为交错型
绝大多数金属茂是交错型构象。
也发现有覆盖型的 , 如 (Me4Cp)2 Ru。
• Al(Me)3+H2O (MeAlO) n (11)
可作为聚合催化剂的金属茂的类型
(M=Ti,Zr,Hf;X=Cl,Br; R=Me)
大多数是柄型夹心化合物 , 是手性分子。这些分子比较刚 性 ,活性空位 ,手性源位置固定且比较接近。
最大的优点是:
结构一致性 , 从而导致催化活性位置的单一 , 这就保证了 催化聚合物的窄分子量分布,这是金属茂聚合物性能上优 于传统催化剂聚合物的原因。
开环夹心化合物又叫开环金属茂,与金属茂比 较 , 其配位体是戊二烯(7)的负离子-戊二烯
大学有机化学第七章 醇、硫醇、酚

C+稳定 脱水成烯越容易, 所以叔醇>仲醇>伯醇。
CH3 b a 提问: CH3-C-CH2CH3 b OH -H2O
*问 140 ℃ H3C C=C H3C
CH3 H
H+ CH3 C
CH3 H CH3 C H CH3 2,3-二甲基-1-丁醇
HOCH2CH2-SH
CH3CH2CH-CH2-CH3 SH
2-巯基乙醇
3-戊硫醇
二、物理性质(略)
三、硫醇的化学性质
1、弱酸性 R—SH + NaOH——>RS-Na+ + H2O 提问: R—OH + NaOH——> X
2、重金属盐的生成
重金属离子:As++、Hg++、Pb++、Cu++、Ag+
3、醛酮与Grignard试剂的加成
+ + H – C = O + R–MgX H2.5 3.5 H 无水乙醚 R-C-OMgX H + MgX2
1° 无水乙醚 CH3-CH-OMgX CH3CHO + CH3CH2MgX CH2CH3 CH3-CH-OH CH2CH3 2° + MgX2
H R-C-OMgX H
与金属钠反应: 伯醇 > 仲醇 > 叔醇
(二)羟基被卤素取代(补充)
R-OH + HX R-X + H2O
反应速度快慢与两种因素有关:
1、与氢卤酸有关: HI > HBr > HCl 2、与醇的类别有关: 如: CH CH3 3 ZnCl2-浓HCl CH3-C-OH CH3-C-Cl + 立即混浊 CH3 CH3
《有机化学》第7章 芳香烃

所以如果希望获得所需的产物,使用正确的反应步骤是重要的。
2023/6/13
18
第三节 稠环芳烃
一、 萘
萘,分子式C10H8,光亮的片状结构,熔点80.2 ℃,沸点218 ℃,有特殊气味,易 升华,不溶于水,易溶于乙醇、乙醚、苯等有机溶剂。其化学性质与苯相似。
2023/6/13
11
⑷ 傅-克(Friedel-Crafts)反应 ① 烷基化反应 凡在有机化合物分子中引入烷基的反应,称为烷基化反应。反
应中提供烷基的试剂叫烷基化剂,它可以是卤代烷、烯烃和醇。
当烷基化剂含有三个或三个以上直链碳原子时,产物发生碳链异构。
② 酰基化反应 凡在有机化合物分子中引入酰基(
①若原有两个取代基不是同一类的,则第三个取代基进入的位置一般受邻、对 位定位基的支配,因为邻、对位基反应的速率大于间位基。
②若原有两个取代基是同一类的,则第三个取代基进入的位置主要受强的定位基 的支配。
2023/6/13
17
⑵ 选择适当的合成路线
例如:由甲苯制备对硝基苯甲酸。
比较这两个结构,反应步骤必须是先硝化,后侧链氧化。
1.取代反应
在萘环上,p电子的离域并不像苯环那样完全平均化,而是在α-碳原子上的电子 云密度较高,β-碳原子上次之,中间共用的两个碳原子上更小,因此亲电取代反应 一般发生在α位。
⑴ 卤化反应
在Fe或FeCl3存在下,将Cl2 通入萘的苯溶液中,主要得到α-氯萘。α-氯萘为无 色液体,沸点259 ℃,可做高沸点溶剂和增塑剂。
2023/6/13
6
苯分子去掉一个氢原子后的基团(C6H5―)叫做苯基,也可以用Ph―代 表。芳烃分子的芳环上去掉一个氢原子后的基团叫做芳基,可用Ar―代表。甲 苯分子中苯环上去掉一个氢原子后所得的基团CH3C6H5―称甲苯基;如果甲苯 的甲基上去掉一个氢原子,C6H5CH2―称苯甲基,又称苄基。
(有机化学课件)第七章 卤代烃

(3) 隔离型卤代烃
卤原子与碳碳重键或苯环相 隔两个或多个饱和碳原子,与 一般卤代烷性质接近。
7.2 卤代烃的命名
普通命名法
简单卤代烃的命名,一般是由烃基的名称加上卤原子的名称而成。
反应
Cl CH2 CH2 OH
Ca(OH)2
CH2 CH2 O
机理
Cl CH2 CH2 OH
HO– – H2O
Cl CH2 CH2 O-
分子内类SN2
CH2 CH2 O
邻基参与 例2
H2O
CH3CH2 S CH2CH2Cl k
CH3CH2 S CH2CH2OH
H2O
CH3CH2 CH2 CH2CH2Cl
亲核试剂的进攻与离去基团的离去同时发生; Nu从L的背后沿着C-L键轴线进攻中心C原子; 中心C原子为手性时,发生Walden 转化,即构型反转。
SN2反应的立体化学特征
SN2反应的立体化学特征为中心C原子的构型反转。
(S)–2–碘辛烷
(R)–2–碘(128I)辛烷
7.6.2 单分子亲核取代反应(SN1)机理
亲核取代反应 (II)
(3) 与氰化钠作用
(4) 与氨作用
C2H5OH (CH3)2CHCH2Cl + 2 NH3
110 oC, 3 h, 84%
(5) 卤离子的交换反应
(CH3)2CHCH2NH2 + NH4Cl 异丁胺
CH3CH CH3 + NaI Br
丙酮 室温
CH3CH CH3 + NaBr I 63%
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Hydrocarbon Functionalization by C-H Activation
C-C bond formation by C-H activation of aromatic molecules
There are a number of examples of C-C bond forming reactions involving C-H activation of aromatic ring C-H bonds. These reactions can even be found in classic organic reactions (Friedel-Crafts alkylation). For a review see: Chem. Rev. 2002, 102, 1731. The metal catalyzed versions of these reactions that couple arenes and alkenes are very attractive because they represent perfectly atom economical reactions (every atom in the starting materials is found in the product).
cat. + 60 + 40
Cat. =
Alkane Functionalization Alkane functionalization reviews: Acc. Chem. Res. 1995, 164. Chem. Rev. 2001, 101, 953-996 J. Chem. Soc., Dalton Trans. 2001, 2437-2450 J. Mol. Cat. A.: Chem. 2004, 230, 7-25
J. Chem. Soc., Chem. Commun. 1982, 1235
J. Am. Chem. Soc. 1987, 109, 8025 t-BuCH=CH2 converts the Ir dihydride to a coordinatively unsaturated Ir(I) complex. The driving force for this reaction is the very exothermic hydrogenation of tBuCH=CH2. The unsaturated Ir(I) species can then oxidatively add the substrate.
• The reaction is not inhibited by CO • Photolysis under 13CO does not result in incorporation of 13CO into the complex • Reaction in pentane and pentane-d12 showed indistinguishable conversion rates, but a large isotope effect was observed in a 1:1 mixture of C5H12 and C5D12 • Photolysis of the W-Bcat' complex in the presence of PMe3 in pentane led to formation of the PMe3 adduct in addition to pentyl-Bcat'. The ratio of PMe3 complex: pentylBcat' was dependent [PMe3] Formation of a strong B-C bond drives this reaction (B-C is 10-15 kcal/mol stronger than C-H, while M-B and M-H have similar bond strengths).
Reactivity of H-CH3 100 X higher than that of HCH2OH Very strong preference for oxidation of C-H bonds on sterically unhindered methyl groups.
For original Shilov chemistry, the Pt(0)/Pt(II) and Pt(II)/Pt(IV) redox couples have similar potentials. Thus Pt(II) can also disproportionate into Pt(0) and Pt(IV). This leads to precipitation of Pt metal and undesired side reactions.
Alkane Dehydrogenation: Alkene hydrogenation is well precedented. Would it be possible to identify systems where the reverse reaction (alkane dehydrogenation) would be favored?
Angew. Chem. Int. Ed. 2001, 40, 3596-3600
Borylation of Alkanes:
1. Stoichiometric Functionalization
(Science, 1997, 277, 211)
Possible Mechanisms:
Observations:
Ph3P
PPh3
Ph3P Ph3P H
Ru PPh3 O H
PPh3
PPh3
R
There are a large number of examples of this type of transformation in which an electrophilic metal center is used to activate a C-H bond on an aromatic center. Typically this occurs by an electrophilic aromatic substitution mechanism.
The Bergman systems are very efficient at oxidatively adding C-H bonds. However efforts to transform the resulting metal-alkyl to a functionalized organic product have met with limited success.
The dehydration can also be carried out thermally without an H2 acceptor if hydrogen can be purged from the reaction system. Catalyst stability becomes an important issue under these conditions (150-230 C). In particular, ligand metallation and other decomposition pathways become important.
H LnM H
R LnM
H H R
H2 H H R + LnMn-2 LnM H H R
The first catalytic system: In order to drive the reaction towards dehydrogenation, it is usually necessary to remove H2 from the reaction system. Often a reactive alkene is used as a hydrogen acceptor. 3,3Dimethyl-1-butene is used as a hydrogen acceptor because of it's high heat of hydrogenation.
Examples where the C-H activation is promoted by coordination to a Lewis-basic functional group are very common.
O + (Ph3P)3(CO)RuH2 Si(OEt)3 toluene, 135 C
Zh. Fiz. Khim. 1969, 43, 2174
CH3OH + HCl
[PtCl4]2-
CH4
H2O
Cl Cl Pt Cl Cl CH3 Cl 2-
HCl 2-
Cl Pt ]2-
[PtCl6]2-
the Shilov system represented a very selective method for alkane oxidation.
C-H activation by transition metals has been demonstrated for numerous systems. Therefore, C-H activation is not such a challenge. However making use of C-H activation to produce functionalized alkanes catalytically still represents a formidable challenge.
J. Am. Chem. Soc. 1982, 104, 352 J. Am. Chem. Soc. 1983, 105, 3929