电子效应和空间效应的教与学
合集下载
有机化学 电子效应和空间效应
C H3C O O H + H2O K1 K2
+ C H3C O O + H3O
C lC H 2 C O O H + H 2 O
+ H3O + C lC H 2 C O O
-
例2 乙醛的水合反应是可逆的,形成的水 化物很不稳定,只能存在于稀水溶液中。而三 氯乙醛的水合反应则比较容易,能生成稳定的 水合物并能离析和长期存在。主要是由于三氯 甲基强烈的-I效应使羰基碳原子带部分正电荷, 亲核反应容易进行,同时水合三氯乙醛因形成 氢键也增加了稳定性。
2.1.7.4 对化学平衡的影响
例1 酸碱的强弱是由其解离平衡常数的大小来衡 量的,在酸碱的分子中引入适当的取代基后,由 于取代基诱导效应的影响,使酸碱离解平衡常数 增大或减小。如乙酸中的一个α-氢原子被氯原子 取代后,由于氯的-I效应,使羧基离解程度加大, 而且使生成的氯乙酸负离子比乙酸负离子稳定, 所以K2>K1 :
2.1.7.2 对反应机理的影响
在一些反应中,由于诱导效应等因素可以改变 其反应机理。如溴代烷的水解反应,伯溴代烷如 CH3—Br主要按 SN2历程进行,而叔溴代烷如
(CH3)3C—Br则主要遵从SN1历程进行。
2.1.7. 3 对反应速率的影响
例1 羰基的亲核加成反应,羰基碳原子的电子 云密度越低,就越容易和亲核试剂发生加成反应, 在这种情况下,分子所需要的活化能就比较小, 容易进入活化状态,因而反应速率较大。故取代 基的-I效应愈强,愈有利于亲核加成;取代基的 +I效应愈强,对亲核加成愈不利。 如下列化合物发生亲核加成的活性顺序为: Cl3C—CHO > Cl2CHCHO > ClCH2CHO > CH3CHO
+ C H3C O O + H3O
C lC H 2 C O O H + H 2 O
+ H3O + C lC H 2 C O O
-
例2 乙醛的水合反应是可逆的,形成的水 化物很不稳定,只能存在于稀水溶液中。而三 氯乙醛的水合反应则比较容易,能生成稳定的 水合物并能离析和长期存在。主要是由于三氯 甲基强烈的-I效应使羰基碳原子带部分正电荷, 亲核反应容易进行,同时水合三氯乙醛因形成 氢键也增加了稳定性。
2.1.7.4 对化学平衡的影响
例1 酸碱的强弱是由其解离平衡常数的大小来衡 量的,在酸碱的分子中引入适当的取代基后,由 于取代基诱导效应的影响,使酸碱离解平衡常数 增大或减小。如乙酸中的一个α-氢原子被氯原子 取代后,由于氯的-I效应,使羧基离解程度加大, 而且使生成的氯乙酸负离子比乙酸负离子稳定, 所以K2>K1 :
2.1.7.2 对反应机理的影响
在一些反应中,由于诱导效应等因素可以改变 其反应机理。如溴代烷的水解反应,伯溴代烷如 CH3—Br主要按 SN2历程进行,而叔溴代烷如
(CH3)3C—Br则主要遵从SN1历程进行。
2.1.7. 3 对反应速率的影响
例1 羰基的亲核加成反应,羰基碳原子的电子 云密度越低,就越容易和亲核试剂发生加成反应, 在这种情况下,分子所需要的活化能就比较小, 容易进入活化状态,因而反应速率较大。故取代 基的-I效应愈强,愈有利于亲核加成;取代基的 +I效应愈强,对亲核加成愈不利。 如下列化合物发生亲核加成的活性顺序为: Cl3C—CHO > Cl2CHCHO > ClCH2CHO > CH3CHO
应用化学系课件第1章电子效应

3、 电子效应和空间效应
• 取代基效应:
• 取代基不同而对分子性质产生的影响。取代 基效应可以分为两大类。一类是电子效应,包括 场效应和诱导效应、共轭效应。电子效应是通过 键的极性传递所表现的分子中原子或基团间的相 互影响,取代基通过影响分子中电子云的分布而 起作用。另一类是空间效应,是由于取代基的大 小和形状引起分子中特殊的张力或阻力的一种效 应,空间效应也对化合物分子的反应性产生一定 影响。
• 又如水与甲醇,均有活泼氢可以形成氢 键:水中的氢键与甲醇中的氢键可以互 相代替,因此水和甲醇可以互溶,但当 醇的分子逐渐增大时,分子中相似部分 即羟基的成分逐渐减少,而不同部分即 碳键的成分逐渐增加,这样水与醇的溶 解度亦逐渐减小。
• 偶极溶剂对于有机试剂或无机试剂溶解度的影 响均很大由于结构上的特点,这种溶剂的偶极 正端藏于分子内部,负端露于分子外部,例如 二甲亚砜:因此它的负端可以与正离子形成离 子一偶极键,正端藏于分子内部,不能与负离 子起作用,故在用负离子作为试剂进行反应时, 由于负离子不被偶极溶剂分子所包围,可以很 容易地进行反应。有些反应在一般的溶剂中进 行比较困难,在偶极溶剂中常常得到比较满意 的结果,速度有时增加很多,因而目前偶极溶 剂的使用日益广泛,成为促进反应速度的一个 重要手段。
• 非离子型化合物分子间.其大小与分子极性有 关,极性越大,偶极-偶极作用也亦愈大,沸点 升高。
• 如果分子内极性相同,则分子愈大,范德瓦引 力亦愈大;故沸点随分子量升高而升高。这一 方面因为分子量增加,分子运动所需的能量增 加,另一方面因为分子增大,分子间的接触面 积增大,即范德瓦引力增大,故怫点升高。
氢键
• 氢键具有饱和性和方向性,健角大都接近于 180o。
• 氢键在很多分子中,起着十分重要的作用,不 仅对一个分子的物理性质及化学性质起着很重 要的作用,而且可以使许多分子保持一定的几 何形象。
电子效应

1,3 - 戊二烯 + H2
254 kj mol -1 E
kj mol -1 226
CH3CH2CH2CH2CH3
说明共轭体系的存在导致体系能量降低
二者之间能量上的差值通称为离域能或共轭
能,它是由于π电子的离域引起的,是共轭效应的
表现,其离域能越大,体系能量越低,化合物则
越稳定。 不对称共轭体系正、负电荷交替传递,并可 沿共轭的碳链一直传递下去,它不因碳链的增长而 减弱。用 表示,起于双键止于与双键相连的 原子(或单键)
H H
(1)σ,π- 超共轭体系: α-碳上C-H σ键与
π键的P轨道可侧面交盖。
σ键的电子偏离原来轨道, 倾向于π轨道
H H
δ
+
H C H C
C H H
H
H CH2 H R
δ
+
C CH H
δ
H
C CH R
δ
+
CH2
δ
C CH R
CH2
δ
C=C更稳定
(2)σ, p - 超共轭体系 能形成σ, p - 超共轭体系的可以是碳正离子或碳自由基
效应通过共轭体系传递的现象,称为共轭效应(C效应)。 共轭体系中必须含有①三个或三个以上②相邻 ③共平面
的原子,这些原子都具有相互平行、且垂直于原子所在平
面的P轨道,可侧面重叠形成多中心、多电子的大π键。
3.产生的结果: ① 电子离域运动.
健长趋向平均化,
电子云趋向平均分布. ②分子内能降低,分子更稳定. ③ 对称体系,电子云平均分布; 不对称体系, 电子云 交替 (δ+ δ– )分布.
一个轨道含S成分:
½ቤተ መጻሕፍቲ ባይዱ
xi第二章化学电子效应与空间效应ppt课件

例如: CH4的偶极矩为零 非极性分子
CH3Cl的偶极矩为 6.24×10-30C.m
1.3诱导效应
诱导效应:在有机分子中引入一原子或基团后,使分 子中成键电子云密度分布发生变化,从而使化学键发生 极化(偏离)的现象,称为诱导效应,又叫I效应。 -I: 吸电子效应 +I:供电子效应
静态诱导效应 由于分子内成键原子的电负性不同 所引起的电子云沿键链按一定方向移动的效应,或者 说键的极性通过键链依次传递的效应。
诱导效应强弱变化规律:
A.同一族的元素随着原子层的增加而吸电子诱导效应降低。 如: —F > —Cl > —Br > —I —OR > —SR —NR2 > —PR2 B.同一周期的元素从左到右吸电子诱导效应增加。如: —F > —OR > —NR2 > —CR3 C.不同杂化状态的碳原子以s轨道成分多者吸电子 能力强。(sp>sp2>sp3) 反映在基团方向时,如:
(C)由核磁共振化学位移粗略比较
X-CH3中甲基的δ值: X δ X -NO2 4.28 -N(CH3)2 -F 4.26 -I -OH 3.47 -COCH3 -Cl 3.05 -COOH -Br 2.68 -CN -SH 2.44 -CH3 C6H5 2.30 H 烷基为吸电子集团 ? δ 2.20 2.16 2.10 2.07 2.00 0.90 0.23
偶极矩的用途:
1.实验测得的偶极矩可以用来判断分子的空间构型。 如同属于AB2型分子,CO2的μ=0,可以判断它是直 线型的;而H2S的μ≠0,可判断它是折线型的。
2.可以用偶极矩表示极性大小。
键偶极矩越大,表示键的极性越大; 分子的偶极矩越大,表示分子的极性越大。 卤代甲烷在气相的偶极矩 CH3 F μ/D 1.82 CH3Cl 1.94 CH3Br CH3I 1.79 1.64
理论有机化学第二章电子效应与空间效应

2. 能量下降 ΔH值:
乙烯(32)
戊二烯1,4(61)
戊二烯1,3(54) 苯(50)
CH3CH2CH2CH3---→ CH2=CH-CH=CH2 只生成1,3-丁二烯, 没有1,2-丁二烯
34
共轭体系和共轭效应
3. 共轭加成 丁二烯的1,4加成
C=C-C=C + H2 → CH-C-C=C → CH-C=C-C+ → CH-CH=C-CH
α,β不饱和醛酮的1,4加成
C=C-C=O + H2 → CH-C=C-OH → CH-CH-C=O
35
共轭体系和共轭效应
共轭插烯作用
R
C
N
O
HN
O
H
双环胺酮无酮或胺的性质,类似酰胺。
36
共轭体系和共轭效应 2.2 静态共轭效应Cs
1.起源与含义:由于共轭双键的存在而 使π电子云发生转移变形,---
CH3
δ-
δ-
δ-
28
§2共轭体系和共轭效应
29
H
§2共轭体系和共轭效应
H
2.P-π共轭:
H H H
由P轨道上的电子与π键组合而成,分四种情况 。
CH2=CH -- P
P轨道有孤对电子
.. CH2=CH-Cl
P轨道有一对电子 P轨道有一个电子
CH2=CH-CH2CH2=CH-CH2.
P轨道上没有电子
推电子 Y→δ-CR3 +Is
H--CR3
吸电子
X←δ+CR3 -Is
2
§1 诱导效应I
eg1 取代酸的酸性变化
CH3COOH CH3CH2COOH ClCH2COOH Cl2CHCOOH Cl3CCOOH
电子效应

电子效应电子效应:取代基不同而对分子性质产生的影响。
取代基效应可以分为两大类。
一类是电子效应,包括场效应和诱导效应、共轭效应。
电子效应是通过键的极性传递所表现的分子中原子或基团间的相互影响,取代基通过影响分子中电子云的分布而起作用。
另一类是空间效应,是由于取代基的大小和形状引起分子中特殊的张力或阻力的一种效应,空间效应也对化合物分子的反应性产生一定影响。
由于取代基的作用而导致的共有电子对沿共价键转移的结果。
诱导效应:当电负性不同的两个原子结合时,共价键就有一定的极性,再多原子分子中,这种极性会通过静电诱导作用而影响到它的相邻部分,使成键电子云偏移到电负性较大部分。
双原子分子:多原子分子:这种由于原子或基团电负性的影响沿着分子中的键传导,引起分子中电子云按一定方向转移或键的极性通过键链依次诱导传递的效应称为诱导效应(inductive effects )或I 效应。
这种效应如果存在于未发生反应的分子中就称为静态诱导效应。
诱导效应的传导是以静电诱导的方式沿着单键或重键传导的,只涉及到电子云密度分布的改变,引起键的极性改变,一般不引起整个分子的电荷转移、价态的变化。
这种影响沿分子链迅速减弱,实际上,经过三个原子之后,诱导效应已很微弱,超过五个原子便没有了。
诱导效应的方向:诱导效应的方向以氢原子作为标准。
-氯代乙酸的酸性。
氯原(位阻效应) 空间效应 取代基效应 空间传递场效应 (σ, π)( π-π, (σ- π,σ- p) 诱导效应 共轭效应 超共轭效应 电子效应 -+++¦Ä¦Ä¦Ä¦Ä¦Ä¦Ä¦ÄC X B AA B C ¦Ä¦Ä¦Ä¦Ä¦Ä¦Ä¦Ä+++-Y CX C H C Y _I ЧӦЧӦI +±È½Ï±ê×¼子取代越多,酸性越强。
电子效应和空间效应
C C
Z
• 特点: • 1、存在于一切分子中,电负性不同引起的。 • 2、是一种静电作用,只涉及到键的极性改 变,不引起电荷转移、变化。而且键的极性 变化一般是单一方向的,即在键链中不产生 极性交替的现象。 • 3、短程传递。超过3个碳已经极弱, 5个碳 便消失了。 • 4、不仅可以沿σ键传递,同样也可以通过π 键传递, • 5、具有加和性
R EtO C H + HBr H
相对速度 1.0 0.28 0.03 4.2× 10-5
SN2 反应: 乙氧基从背后进攻,
R越大,位阻越大
(CH3)3C(t-Butyl)
•
另一类空间效应是由于张力而引起的,空间效应 也并不都是阻碍反应进行的,有的是促进反应进 行的。例如卤代烷按SN1历程进行的水解反应, 由于反应物卤代烷是四面体结构,中心碳原子为 杂化状态,键角接近109.5,而活性中间体碳正离 子为平面结构,中心碳原子为杂化状态,键角为 120,由变为,原子或基团之间的空间张力变小, 容易形成,而且原子或基团的体积愈大时,杂化 状态下张力也愈大,转变为杂化状态张力松弛地 也愈明显,形成碳正离子愈容易,碳正离子也愈 稳定。
•
•
• •
•
如碳-卤键的极性次序应为:C—F>C—Cl>C— Br>C—I 但卤代烷的亲核取代反应活性却恰恰相反,实际 其相对活性为: R—F<R—Cl<R—Br<R—I 原因就是动态诱导效应的影响。因为在同族元素 中,随原子序数的增大电负性降低,其电子云受 到核的约束也相应减弱,所以极化性增大,反应 活性增加。如果中心原子是同周期元素,则动态 诱导效应次序随原子序数的增大而减弱,因为电 负性增大使电子云受到核的约束相应加强,所以 极化性减弱,反应活性降低。-CR3>-NR2>-OR>F 相同元素具有电荷时则必为这样顺序:-O—>OR>-
第三章 电子效应和空间效应
例: CH3δ+ Clδ键距 μ=qd q: 中心电荷 d:正负电荷中心的距离
分子的偶极距是各键的键距向量和:
H H H
μ=0
Cl H Cl Cl
μ=0
Cl Cl H H
μ=1.94D
C
C
C H
Cl-Cl ( 键距为零)
3.2诱导效应
4.2.1 定义:诱导效应是指在有机化合物中由于电负性不同的取 代基的影响,使整个分子中成键电子云按取代基团的电负性所决定 的方向而偏移的效应。
-I效应:
CH3δδδ+-CH2δδ+-CH2δ+→Clδ-
+I效应:
O CH 3 C H
诱导效应在没有外加电场影响下也存在,它体现的是分子自身的性质。诱
导效应一般用 I 表示,饱和 C-H 键的诱导效应规定为零。
当一个原子或原子团与碳原子成键后,电子云偏离碳 原子,称为-I 效应。 例:
O H3C X H3C H H3C
非极性共价键:相同原子(基团)成键,电 子云分布对称 极性共价键:不同原子(基团)成键,电子 云分布偏向 共价键极性:取决成键原子的相对电负 性.是结构与反应性能关系的基础
极性共价键: 形成共价键的原子,它们之间吸引电子 的能力是不一样的。这就使得两原子间 共价键的电子云不是平均分配在两个原 子核之间,而是偏向电负性较大的原子, 这种键成称为极性共价键。
•
3.4.2 对反应机理的影响
在一些反应中,由于诱导效应等因素可以改变其反 应机理。如溴代烷的水解反应,伯溴代烷如CH3—Br主 要按 SN2历程进行,而叔溴代烷如(CH3)3C—Br则主要 遵从SN1历程进行。
3.4.3 对反应速率的影响
1第一章 有机化学中的电子效应和空间效应10
2021/7/9
13
4、取代基诱导效应的相对强度测定方法: • 由取代酸碱的解离常数确定 • 比较分子的偶极矩 • 比较核磁共振(NMR)的化学位移 • 比较诱导效应指数
2021/7/9
14
5、取代基诱导效应的应用
2021/7/9
15
2021/7/9
16
三、 共轭效应 (Conjugative Effect)
H CH3 H
2021/7/9
9
二、诱导效应(Inductive Effect) 1、概念:由于成键原子的电负性不同,而使整个分子的电子云沿着碳链向某
一方向移动的现象。
2、性质: 1)静态诱导效应是由键的极性和分子结构决定的一种永久性效应。 2)诱导效应是一种短程效应,沿原子链由近而远依次传递,并愈来
4.08
4.09
4.20
③ 邻位取代芳香酸的酸性都较苯甲酸的酸性强。
COOH
COOH
C(CH3)3 >
2021/7/9
C(CH3)3
40
COOH NO2
pKa 2.17
COOH NO2
3.42
COOH
NO2
3.45
④ 如果邻位基团能与羧基形成氢键,使酸性增强。
COOH a.
COOH b.
OH
COOH
东理工大学出版社。 4. 《高等有机化学》,[美]J.马奇著,高等教育出版社。
2021/7/9
2
2021/7/9
3
引言
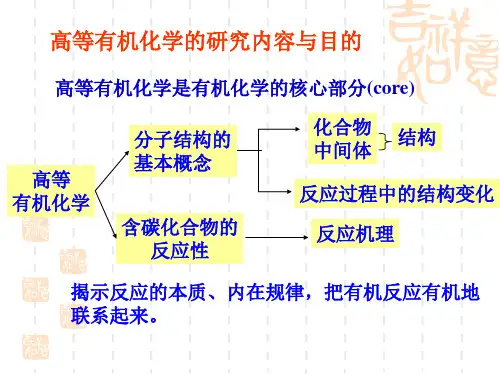
高等有机化学又名物理有机和理论有机化学,是一门论 述有机化合物的结构、反应、机理及它们之间关系的科 学,对整个有机化学起着理论指导作用。
研究对象:有机化合物的结构以及有机化合物在反应过 程中结构的变化,研究有机分子的结构和反应条件对有 机化合物的物理、化学性能的影响以及化学反应历程。
第二章 电子效应和空间效应
简单分子的弹力常数可由分子振动光谱分析结果计算。 -----它代表着键的伸长、缩短及张合的难易程度。
利用振动频率和形成共价键的原子的质量,可根据下列关系式得到其近似值。
f
2 M
振动频率 f: 弹力常数 M: 折合质量
1
1
1
=
+
M
m1
m2
单键 双键 三键
7 x 10-5 7 x 10-5 - 15 x 10-5 15 x 10-5
一:共价键的极性和极化性
1. 共价键的极性 由于分子中成键原子的电负性不同,使成键原子间电子云 分布不均,共价键具有一定偶极矩的现象。
电负性:不同元素原子吸引电子的能力,也可以认为是成键原子对共享电子对 吸引能力。
键的极性指数: 描述键的极性大小的指数。
1)电负性差(电负性和)
K-Br Li-Cl Al-O B-F
第二章 电子效应和空间效应
学习目的:对电子效应的产生、种类、相对强度及其对有机分子
化学行为的影响有一个系统的认识,掌握一般规律; 对空间效应通过实例介绍空间张力、空间障碍对化学 反应的影响
1. 诱导效应 2.共轭效应 3.超共轭效应 4.同共轭效应 5.空间效应
概述:当有机化合物分子相互作用形成活化络合物时,不管是新键的形成还是 原来旧键的破裂,都直接或间接地与分子中电子云的分布有关。所以通过对 电子云变化情况的研究,可以来解释或预测有机化学中的很多问题。
B: 原子半径 有效荷电量相同时,原子系数增大,键的极化性增大。 C—X 键的极化性:C—I > C—Br > C—Cl > C—F
C: 键型
>s
3.极性和极化性的量度-偶极矩
偶极矩(Bond Dipole Moment)包括键的偶极矩简称键矩和分子偶极矩。 它是衡量化学键与分子极性的物理量。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
收稿日期: 2006—12—09 作者简介: 张淑萍( 1963— ) , 女, 山西阳城人, 讲师, 主要从事有机化学教学与研究。
· 63 ·
长治学院学报
2.1.3 键 长 氟 代 烃 C- F 键 长 的 变 化 是 由 于 氟 原 电子。此外, 甲基的 3 个 C- H 键的 σ 电子和苯环形
超共轭效应。空间效应在有机化学中相当普遍, 当分 化学性质也被有机联系在一起不必死记硬背而易于
子内的原子或基团处于范德华半径不许可的范围时 学习掌握了。
产生的排斥作用, 或两个分子相互接近时由于基团 2 应用分析
之间的非键作用所引起的化学效应, 都是空间效应
下面通过一些实例, 从多个不同的侧面概略地
有机化学教学中, 同学经常向老师提的问题是: 如何 的影响, 以及反应速率、平衡、位置及产物等的影响。
才能学好有机化学? 我总会提请同学注意掌握电子 一般来讲, 有机化合物官能团的共性较清楚明了, 规
效应。
律性强, 易于掌握, 难点在于分子中不同的取代基对
1 电子效应和空间效应及其重要作用
官能团影响所反映出的众多差异性和特殊性。因此,
甲基的碳原子是 sp3 杂化, 苯环的碳原子是 sp2
··
如苯环上带有含氧、氮原子的基团, 如: - N HCHO, -
··
··
··
··
··
··
··
O COR, - N HCOR, - O R, - N H2, - N HR, - N HR, - N
HR2 等, 它们与碳原子既有 p- π 的 +C 共轭效应,
电子效应来解释:
HX F3CCH=CH2
+
F3CC H- CH3
( Ⅲ)
+
F3CCH2- C H2
( Ⅳ)
F3CCHX- CH3 ( 次) F3CCH2- CH2X ( 主)
由于 F3C- 的强吸电子作用,( Ⅳ) 式的稳定性强 容易进行。 于( Ⅲ) 式, 过渡态结构稳定, 势能低, 活化能小, 反应 2.3.3 醇酸性消除
不对称烯烃加成有两种可能的取向产物。电子 效应分析, 中间正碳离子的稳定性为:
+
+
+
+
+
CH2=CHC H2> R3C > R2C H> RC H2> C H3
因而大多数都符合马氏规则。但双键碳上带有强吸
电子原子或基团, 如: - CX3、- CN、- COOH 等, 则主要 生成反马氏规则产物, 但仍符合电性规律, 也可以用
也有 - I 的诱导效应。由于孤电子对的 p 轨道与碳的
p 轨道在同一能级, 故 +C﹥- I, 而电负性 O 原子﹥N
原子, 苯环上的电子云密度逐渐增加, 亲电取代反应
速率逐渐加快。对芳卤而言( 除氟外) , 卤原子与环既
有 p- π 的 +C 共轭效应, 也有 - I 的诱导效应。由于
卤原子孤对电子的 p 轨道与碳原子的 p 轨道不在同
参与共轭, 由于 N 的电负性比较大而相对于吸电子 子或基团时, 其电子云密度大大降低, 而多采取亲核
基 团 , 亲 电 取 代 一 般 发 生 在 β- 位 , 亲 核 反 应 则 常 加成反应历程, 如 F2C=CF2、( NC) 2=( CN) 2 等。同样, 发生在 α- 位; N- 氧化的三级胺, 由于其碱性较强, 对于碳氧双键而言, 由于氧的电负性大, 增加了碳原
2007 年 4 月 第 24 卷 第 2 期
长治学院学报 Journal of Changzhi University
Apr. ,2007 Vol.24,No.2
电子效应和空间效应的教与学
张淑萍
( 长治学院生化系, 山西长治 046011)
摘 要: 诱导效应、共轭效应和超共轭效应总称为电子效应, 它是有机物结构和性质的重要的理论依据。该文简单地分
子的 p 轨道与碳原子上 π* 型轨道发生超共轭作用 成了 σ- π 共轭体系, 这个 σ- π 共轭效应也使苯
造成的, 也可用立体电子效应来解释[2]。
环活化。因此, 诱导效应和超共轭效应都使苯环的电
2.2 反应活性与速率
子云密度增大。故甲苯比苯易于发生亲电取代反应。
2.2.1 芳烃的亲电取代 苯发生硝化反应温度为 55 ̄60℃, 硝基苯硝化
电子效应是分子中某些原子或基团所引起的诱 可采用这样的程序学习各系列化合物: 在掌握官能
导效应、共轭效应和超共轭效应的总称。多原子分子 团一般特性的基础上, 利用电子效应搞清各化合物
中一个键产生的极性将影响到分子的其余部分, 电 的电子云密度分布特征, 并结合空间效应综合分析
子的转移可以按静电诱导方式沿分子链传递, 也可 其对官能团性质的影响及其影响的大小, 以把握不
由于烷基与双键的超共轭作用, 过渡态( Ⅵ) 式 应为顺式消除, 主要就是由于空间效应决定的。
的稳定性强于( Ⅴ) 式, 势能低, 活化能小, 反应容易 2.4 反应类型
进行。该产物属热力学控制产物。
2.4.1 亲电加成变为亲核加成
五元杂环化合物, 如呋喃、噻酚、吡咯中的 O、S、
对于烯烃, 碳碳双键的电子云密度较大且易暴
N 等原子, 由于有一对孤对电子参与到苯环共轭体 露, 易于发生亲电加成反应。当双键碳原子上连有给
系中, 相当于有推电子基团, 亲电取代反应主要发生 电子基时, 可加速亲电加成反应。当连有吸电子基
在 α- 位; 而吡啶六元环中的 N 并不提供孤对电子 时, 亲电加成反应变得困难。而当连有多个吸电子原
·· ·· ·· ··
··
甲基的碳原子是 sp3 杂化, 烯碳原子是 sp2 杂化, 从 OR, OH, NH2, OHR, OR2 等, 它们与烯碳原子之间的
轨 道 电 负 性 看 sp2﹥sp3, 所 以 , 甲 基 表 现 为 供 电 子 。 效应与苯环相似。烯键上的电子云密度逐渐增加, 亲
此外, 甲基的 3 个 C- H 键的 σ电子和双键形成了 电加成反应速率逐渐加快。烯卤( 除氟外) 与芳卤相
· 64 ·
张淑萍 电子效应和空间效应在有机化学学习中的作用
烃。该反应称为 Hoffmann 消除( 即反 Saytzeff) 反应, 2.3 反应产物取向
属动力学控制。而 Saytzeff 消除反应, 属热力学控制。 2.3.1 烷烃卤代
空间效应有时甚至能阻止看起来容易进行的反应,
当烷烃分子中有两种以上不同类型的氢原子
析了电子效应, 并列举电子效应的一些应用。
关键词: 电子效应; 空间效应
中图分类号: O621
文献标识码: A 文章编号: 1673- 2014( 2007) 02- 0063- 04
有机化学的初学者对数量具大、种类繁多的有 包括有机化合物的物理性质、反应活性、中间体稳定
机化合物以及复杂多样的化学反应而不知所措。在 性、酸碱性、对亲电、亲核和自由基等类型反应取向
温度需 95℃, 而甲苯硝化温度仅仅需 30℃。这是因 为: 硝基与苯环共平面, 构成了共轭体系, 氮和氧的 电负性又很大, 故使共轭体系的电子云移向硝基, 降 低了苯环的电子云密度, 其中以邻位和对位为甚, 而 间位相对来说还是较高一些。对于其它一些取代基 如 : - COR, - CHO, - CO2R, - CONH2, - COOH, - SO3H, - CN, - CX3, +NR3 等, 除 - CX3 只有 - I 的诱导效应外, 其它基团既有 - I 的诱导效应, 也有 - C 共轭效应, 且效应方向一致, 对苯环的致钝作用逐渐加强, 间位 定位作用也在逐渐加强。
团的性质, 例如: 氯苯很难进行亲核取代反应, 但 烃基 R 的推电子作用。
2,4,6- 三硝基氯苯在室温下就可与 NaHCO3 溶液发 2.1.2 沸点 abC=Cab 型烯烃 ( a,b 为任意取代基)
生亲核取代[1]。
的顺式异构体总是偶极分子, 沸点也比无偶极矩的
电子效应的影响涉及到有机化学的很多方面, 反式异构体要高。
一小时后反应产率:
!HCHO
RCHO 70 ̄90%
=0 CH3COCH3 CH3COCH2CH2CH3 CH3CH2COCH2CH3 PhCOCH3
35%
22%
12%
2%
1%
这是因为: 随着烃基 R 体积的增大, 增加了与 负性很大, 羰基对苯环的诱导效应和共轭效应都使 亲核试剂的排斥力, 使它难于接近羰基碳原子。这 苯环的电子云偏向羰基, 减弱了羰基碳原子的亲电 也说明了为什么醛比酮的加成要容易得多。另外, 性。总之, 醛、酮的结构对羰基亲核加成反应活性 在 反 应 过 程 中 , 羰 基 碳 原 子 是 从 sp2 杂 化 变 为 sp3 的影响是电子效应和空间效应 ( 包括环状物的角张 杂化的。R- C- R′键角是变小的, 若 Nu、R、R′体 力) 综合作用的结果[4]。 积 增 大 , 则 不 论 分 子 内 的 化 学 键 间 或 Nu、R、R′ 另 外 , 卤 代 烷 的 亲 核 取 代 反 应 不 但 反 应 机 理 间的排斥力都增加了。可见, 空间因素对亲核加成 ( SN1 或 SN2) 主要取决于分子结构, 且其相对反应速 有着重大影响。另一方面, 烃基 R 碳原子是 sp3 杂 度亦决定于电子效应[5]。而不同卤素的相对活性( RI 化, 而羰基碳原子是 sp2 杂化, 从轨道电负性看 sp2 ﹥RBr﹥RCl) 则是由于动态诱导效应造成的结果[6]。 ﹥sp3, 所 以 , 烃 基 表 现 为 供 电 子 作 用 , 从 而 消 弱 彻底甲基化的季铵盐, 转化成季铵碱后显较强碱性, 了羰基碳原子的亲电性。对于苯乙酮而言, 氧的电 略显酸性的 β- H, 在加热下容易被夺去而生成烯
略显酸性的 β- H, 则容易被夺去而生成烯烃。该反 子的亲电性, 羰基易于进行亲核加成。