高等有机化学 第3章_有机化学反应机理研究讲解
《高等有机化学—反应和机理》(Bernard Miller)笔记

●Woodward-Hoffmann规则一:4n电子的热电环化反应,如果按照顺旋方式进行是允许的;4n+2电子的热电环化反应,如果按照对旋的方式进行时允许的。
●顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以相同方向(同为顺时针或同为逆时针)旋转成键或断键,这种方式称为顺旋。
顺旋:多烯烃的末端碳原子或环烯烃的饱和碳原子,以不相同方向旋转成键或断键,这种方式称为对旋。
●最高已占分子轨道(HOMO)在4n+2的体系中是对称的;最低未占分子轨道(LUMO)在4n+2的体系中是反对称的。
●前线轨道理论:忽略较低的能级轨道,只考虑HOMO。
前线轨道理论能简单、形象化,但是理论上不完善,在理论上应该有更精确的处理方法。
在电环化反应中,对旋是允许的,顺旋是禁阻的。
●轨道对称性守恒:反应物中的每个轨道的对称性,在反应后对称性保持不变。
●用相关图法处理电环化反应遵循轨道对称性守恒。
●相关图法处理4n+2体系的热环化反应(对旋):以1,3,5-己三烯为例:(1)形成6个分子轨道(2)用能量最低的形成键,和的对称性相同,都是镜面对称的。
(3)是由6个原子轨道组成,键是2个原子轨道组成,故转化为时,可以想象其中有4个原子轨道的系数降低为0。
(4) 1,3,5-己三烯的,不能转化为1,3环己二烯的,因为前者的的对称性是镜面反对称,后者的的对称性是镜面对称,对称性不匹配。
故1,3,5-己三烯的事转化为1,3环己二烯的,同理1,3,5-己三烯的事转化为1,3环己二烯的(5)能量分配很合理,故反应是允许的。
用相关图法处理4n体系的热环化反应(对旋):以1,3-丁二烯为例:(1)用能量最低的形成键(2)用1,3-丁二烯的形成环丁烯的;用1,3-丁二烯的形成环丁烯的。
理由同4n+2体系,因为对称性不守恒。
(3) 1,3-丁二烯的上有2个电子,而要形成的环丁烯的电子在上。
但是1,3-丁二烯要转化为环丁烯的,如果发生这样的转化,就会形成能量很高的环丁烯的激发态。
高等有机化学第三部分3-3.ppt
R-X + Y
. . R X
R +Y
R-X + Y(自由基离子形成)
.R + X
(电子转移)
RY
. or: R + Y-
RY
R Y +R X
R Y+ R X
10
SET的例证之一
.
Y
X +Y
.
(X=I, NO2等)
11
(四) 邻基参与机理(Neighboring-Group) 常有这样的情况: a.反应速度比预期的快 b.构型保留
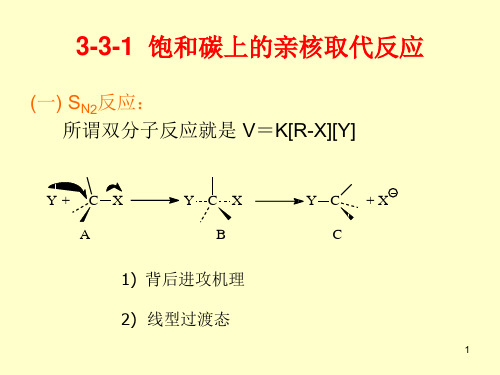
3-3-1 饱和碳上的亲核取代反应
(一) SN2反应: 所谓双分子反应就是 V=K[R-X][Y]
Y+ C X A
YCX B
Y C +X C
1) 背后进攻机理 2) 线型过渡态
1
1) 背后进攻机理
虽然SN2机理是在1937年由Hughs和Ingold 等人正式提出,但早在1893年Walden就发现了 这个取代反应中的构型翻转,所以现在人们又 称Walden翻转。
RX
RX
RX
R +X
紧密
松散
游离的离子
部分构型保留
消旋化
(溶液分隔)
部分保留和彻底的消旋化是SN1的特点。
注意:实际上很少纯粹的SN1或SN2反应,很多是二者都有。
9
(三)SET机理(Single Electron Transfer) 在亲核取代反应中有一类含有自由基或自由基
离子中间体,这类反应是通过SET机理进行的。
Allyl
40
Bn
120
* 位阻小的反应快,烯丙基反应快。
25
高等有机化学 第三章 亲核取代反应(2010)

31
R
CH3CH2 CH2=CHCH2
C6H5CH2
相对速度: 1
40
120
⑤ -C是桥头碳,分子的立体结构使亲核试
剂没有从背面进攻的可能,所以这样的化
合物不能按SN2反应历程进行反应。
L
32
(2)离去基团(L)的性质
一般来说,离去基团越容易离去,
SN2越快。反应时,L是带着原来与
相对速度: 1
410-1
310-2
110-5
其中,新戊基与异丁基相比,SN2的速度
大大下降,这也是位阻的结果。
28
② -C是不饱和碳,SN2很难发生。如乙烯基
卤化物和芳基卤化物都很难水解。
.. C=CH X
p- 共轭效应,X 很难离去
CH2=CH X
X
很难水解!
29
③ 若-C上连有C=O、CN等吸电子基团,
RX + Y:
例如
RY + X:
-
NaI CH3CHCH2CN acetone CH3CHCH2CN
Br
I
11
LiBr CH2OTs acetone
CH2Br
CH3CH(CH2)5CH3 + PhS - ethanol CH3CH(CH2)5CH3 SPh OTs
12
(3)正离子底物 + 中性亲核体
的条件。这种条件变化使负离子稳定性产
生差异,对于不同的卤素,影响程度是不
同的。卤原子愈小,影响程度愈大。所以 RCl、RBr 和RI在DMF中的相对活性差距, 与醇水溶液中的差距相比就大了。
38
(3)亲核试剂性质的影响
亲核试剂的亲核性愈强,浓度愈高,反
有机化学反应机理的研究

-0.27
-0.01 -0.17 0
I
COMe CF3 NO2
+0.352
+0.376 +0.43 +0.71
+0.276
+0.502 +0.54 +0.778
由表可知,凡σ为正值的原子团都是吸引电子的原子团,而 σ为负值的原子团都是排斥电子的原子团。
2、反应常数ρ
取代基常数σ定量地表示取代基的极性效应,只 取决于取代基的结构和位置,与反应类型和反应条 件无关。 • 研究取代苯甲酸酯的水解反应,建立了关系方程 哈密特(Hammett)方程: lgK/K0 = σρ 或 lgk/k0 = σρ K和K0、k/k0分别为取代苯甲酸酯和苯甲酸酯的水 解平衡常数和速率常数 • ρ称为反应常数,是反应对取代基极性效应的敏感 性的定量尺度,ρ决定于反应特性和条件,同一类 型的反应在相同条件下ρ值相同。
五、立体化学证据 从立体化学的信息获得有关机理的证明 根据化合物的构型、构象等变化作出理性的判断
空阻差别不大, 得热力学产物
樟脑衍生物 分子中羰基 的还原
六、动力学证据
• 研究各种反应因素,如浓度、压力、温度、催化剂等对 反应速度的影响。可以提供有关反应过程的信息。 • 反应速度是反应物消失的速度或产物生成的速度 。多步 反应中,决速步骤是反应速度最慢的一步。
第二节 热力学控制及动力学控制
某一反应物A在一定条件下可能转变成两种产物B和C
速率常数 kB<kC (形成C快于B)
平衡常数 KB>KC (B比C更稳定) B
A
C
1、反应初期
⊿G C <⊿GB (活化自由能) kC>kB , A转变成C较容易, [C]>[B] 2、若平衡 主产物为C
《有机反应机理》PPT课件

精选ppt
有机反应机理 22
热力学控制
当产物A能以一定的途径转变成产物B或生成 A和B的反应为可逆反应时,最终产物将受热力学 控制
精选ppt
有机反应机理 23
2.5.5 Hammond假设和过渡状态结构
过渡状态结构决定了:
反应速率 取代基效应 溶剂效应
精选ppt
有机反应机理 24
由于过渡状态寿命短,难以分离或检测,主要通过 以下一些途径间接推测过渡态的结构
例如,1,1’-偶氮丁烷的气相热分解, S=79.5J· mol-1·K-1 >0,表明C-N键的断裂 是 反应的速率控制步骤
B uNNB u B u NNB u
B u+N 2
精选ppt
有机反应机理 42
又如,叔丁基过氧化物的热分解, S =57.5J·mol-1·K-1,与以下机理相一致
(C H 3 )3 C OO C (C H 3 )3
解释有机化学中的一些基本定理,如
Markownikoff规则。 (2.5)
精选ppt
有机反应机理 2
2.5 与过渡状态理论有关的概念或推论
2.5.1 反应的自由能图与活泼中间体
根据过渡态理论,反应速率的快慢与反应的活化 自由能的大小有关
活化自由能是过渡态与反应物的自由能之差,通常 用反应的自由能图来表示反应过程中自由能的变化
精选ppt
有机反应机理 7
两步反应的自由能图与活泼中间体:
G
TS1 TS2
Int
R
P
q
精选ppt
有机反应机理 8
2.5.2 微观可逆性原理
文字描述:
相同条件下,正逆反应具有相同的反应途径
微观可逆性原理是过渡状态理论的必然推论,因为 相同条件下,体系的势能面是相同的,正逆反应的 最低能量途径必然也是相同的
有机化学反应的动力学和机理 ppt课件

虽然设想及判断反应机理是否合理,似乎靠
经验或直觉,但还是有一定的基本规则。
ppt课件
35
确定反应机理的基本规则
(1)简要原则(Simplicity Principle) 机理应尽可能简单,但能够解释全部实验
事实。 如果有几种假设或设想与实验事实都相符
合,则选择最简单的一个。
ppt课件
36
碳正离子、碳负离子、卡宾等,可利 用IR、NMR、ESR等技术检测其存在。
ppt课件
大多数化学家认为:
☛ 化学的真正核心就是研究反应机理,即测 定分子进行化学反应所发生的一些具体的 变化和相互作用。
☛ 如果完全描述一个有机反应的机理,必须
知道反应物分子转变为产物的全过程中所
有原子在不同时间的确切位置。
ppt课件
3
☛ 这是一个从来也没有完全实现过的目标! ☛ 因为许多变化对于任何能直接监测的方
ppt课件
22
一些重要的基本概念:
(1)反应机理(Reaction Mechanism)— —通过一系列基元反应来实现的化学变 化的详细过程。
(2)基元反应(Elementary Reaction)— —只有一个过渡态而不包含任何中间体 的过程。
ppt课件
23
一些重要的基本概念:
(3)过渡态(Transition State)——在基元 反应过程中经历的具有最高势能的结构。
而过渡态发生在10-13s内完成。假设整个过 程放大1013倍。即过渡态在1s内完成,反应 的时间则为200万年。
ppt课件
27
600 1013 200 万年 365 24 60 60
目前直接用实验的方法研究过渡态是极为
第三章__有机化学反应

诱导效应的影响。
诱导效应不仅可以沿σ键链传递,同
样也可以通过 π键传递,而且由于π键电
子云流动性较大,因此不饱和键能更有效
地传递这种原子之间的相互影响。
②动态诱导效应。 在化学反应中,当进攻试剂接近时, 因外界电场的影响,也会使共价键上电子 云分布发生改变,键的极性发生变化,这 被称为动态诱导效应,也称可极化性,用 Id表示。
• • • •
3.2.1 亲核取代反应机理
此反应是亲核试剂对带有部分正电荷的碳原子进
行攻击,属于亲核取代反应,用 SN 表示。 R-L 是受攻 击对象,称为底物;把进行反应的碳原子称为中心碳
发生动态导效应时,外电场的方向 将决定键内电子云偏离方向。如果Id和Is 的作用方向一致时,将有助于化学反应
的进行。在两者的作用方向不一致时,
Id往往起主导作用。
③诱导效应的相对强度
对于静态诱导效应,其强度取决于原子或
基团的电负性。
( a )同周期的元素中,其电负性和 -Is 随族数的 增大而递增,但+Is则相反。如: -Is:—F>—OH>—NH2>—CH3
亲核取代反应的类型 常见的亲核取代有以下四种类型: 1)中性底物与中性亲核试剂作用; R—L + Nu: → R—Nu+ + L2)中性底物与带负电荷的亲核试剂作用; R—L + Nu:- → R—Nu + L-: 3)带正电底物与中性试剂作用; RL+ + Nu: → RNu+ + L: 4)带正电底物与带负电亲核试剂作用; RL+ + Nu:- → R—Nu + L:
3.2 亲核取代反应 (Nucleophilic Substitution )
《有机反应机理》PPT课件

第1章学习要点
1 什么是反应机理?研究反应机理有何意义? 2 研究反应机理的步骤和方法 3 如何表示反应机理? 4 共振结构,酸碱理论等概念 5 重要离子型反应的机理
1.1 反应机理的定义和研究机理的方法
1.1.1 定义 理想的机理模型: 建立体系所有原子空间位置与时间的函数关系, 可以求任意时刻体系中所有原子的空间位置
哪一步是速率控制步骤(rate determining step, rds)
反应条件对反应速率有何影响
可以认为
反应机理是对组成一个化学反应的全部基元过程、 全部中间体(Int.)以及与中间体、反应物(R)和 产物(P)相关联的过渡态(TS)的总体描述
以上也可看作是反应机理的定义
1.1.2 研究机理的步骤和方法 研究机理的步骤
研究机理的方法
动力学方法-速率 速率与温度的关系-活化参数 速率与浓度的关系-动力学方程
同位素的应用 动力学同位素效应 平衡同位素效应 同位素标记
活性中间体的研究 立体化学的研究 密切相关体系的研究
1.2 反应机理的分类与表示法 1.2.1 有机反应与机理的分类 1.2.2 有机反应机理的表示法
自由基( free-radical)反应 反应中伴随单电子转移过程 自由机反应可能为链反应,也可能为非链反应
周环( pericyclic)反应 经由环状过渡态的协同反应
金属催化(metal-catalyzed)反应
由过渡金属催化或促进的各种反应
有些过渡金属 (如 TiCl4, FeCl3) 在反应中仅起Lewis 酸的作用;有些金属在反应中仅提供电子(如Na 和Li) 这些金属参与的反应一般分类为极性反应,周环 反应或自由基反应
IUPAC表示机Biblioteka 的符号及意义见下表符号 A D + * E N R e n r H h xh C P int SS {}
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
二、路易斯酸碱定义
1、概念: 凡是能接受外来电子对的叫路易斯酸;能给予电子对的叫路易 斯碱。 2、与有机反应试剂性质的关系: 路易斯碱一般是亲核试剂;路易斯酸一般是亲电试剂
CH3CH2COO-H + OHCH3CH2O-H + OH-
CH3CH2-Cl + OHCH3CH2-OH + ClCH3CH2-OH2+ + Cl-
3、与布伦斯特酸碱概念的区别: 路易斯定义碱一般就是布伦斯特定义碱;但酸的概念不同。
NH3 、OH- 、既是路易斯碱也是布伦斯特碱 HCl、H2SO4是属于布伦斯特定义的酸,但只有它的H+才是 路易斯定义的酸。
三、酸碱强弱与酸碱反应
1、酸强度的表示法:
HA + H2O
H3O+ + A
*Ka:值大,酸性强,否则相反。
取代
结论:形成烯醇化物是控制速率步骤。
3、溶剂同位素效应 定义:溶剂由H2O 变为 D2O ;ROH变为 ROD时,
反应速率发生变化的作用
2H2O 2D2O
H3O+ + HOD3O+ + HO-
kH / kD =6.5
说明:两者的电离程度不同,水大于重水。 水碱性大于重水,络合正离子能力大于重水。
例如:对于下列反应,实际上还有少量乙烷生成:
CH4 + Cl2 hv CH3Cl + CH3CH3
若提出的机理不能解释少量乙烷在这个反应中形成的 原因,那么这个机理不可能是正确的。
(CH3. + CH3.
CH3-CH3 )
二、中间体的确定: 确定中间体是否存在,确定中间体结构的方法有以下 几种
1、将中间体分离出来 2、用四大谱检测中间体的存在和相应结构 3、中间体的捕获(加入捕获剂,使其与中间体
3-1有机反应机理分类
一、机理 分类
根据键断裂的方式: 或进攻试剂的性质
离子型反应 自由基历程
周环反应
根据反应物和生成物的关系
取代反应 加成反应 消去反应 重排反应
二、试剂分类
亲核试剂: 带负电荷、带有孤对电子的、富电子的、路易斯碱
HO- . RO- CN- ROH R3N H- -C=CR
亲电试剂:带正电荷 、缺电子的、路易斯酸 H+ . Br+ . +NO2 . SO3 BF3 B2H6 Hg(OAc)2
第三章 有机反应机理的研究
§ 3-1 有机反应机理类型 § 3-2 确定有机反应机理的方法 § 3-3 动力学和热力学控制 § 3-4 取代基效应和线性自由能关系 § 3-5 有机酸碱
概述 反应机理是研究反应发生的实际过程。它包括哪些键断裂和生 成,以什么样的顺序进行。有几步反应,它的立体化学要求如 何?反应结合点在在哪里?哪些因素会影响反应的速度? 研究反应机理能帮助我们把许多表面看起来毫不相关的反应联 系在一起,找出共同的规律,用以指导反应条件的选择,达到 提高产率的目的,并为合成新化合物提供理论依据。
2)溶剂化原理:能结合正离子,但不能溶剂化负离子, 负离子是裸露的,有利于SN2反应。
例:在0oC下、CH3I和N3-离子的反应速度常数 在甲醇溶剂中:1 在二甲基亚砜中:45X104
(因它们只能与正性部份或正离子进行溶剂化;由于
空阻作用,不能与负离子进行溶剂化作用。因此负离
子是裸露的,进攻性特强.) O- H H
3-2热力学控制与动力学控制
二、热力学控制反应: 以产物稳定性取胜
一、动力学控制反应: 以活化能低取胜
CH2 CH CH CH2 HCl CH3 CH CH CH2Cl + CH3 CH CH CH2
1,4-加成
Cl 1,2-加成
(-80oC) 20%
80%
(20oC) 80%
20%
三、反应能量曲线:(见下页) 四、常见:萘的磺化反应、苯的烷基化反应。
lg k
k0
lg K , because 1, so lg K
K0
K0
:取代基常数,反映了取代基对苯甲酸电离常数的影响
:反映了反应对取代基效应的敏感程度。同类反应为定值。
四、通过线性自由能关系式测定σ值
若σ值为正,说明取代苯甲酸的电离常数大于苯甲酸,该取代基 为吸电基; σ值越大,吸电能力越强。
如不能分离,也不能捕获, 也可以用检测的办法. 现在可 以用NMR, IR, Raman, 顺磁共振等光谱做反应的实时检测. 甚至要求很高的无水无氧的金属有机反应也可以在核磁管中 做. 以前例如用Raman光谱检出NO2+的存在, 是苯硝化反应中 有此中间体的强有力的证明.
HNO3 + 2H2SO4 2HNO3
三、质子极性溶剂和非质子极性溶剂
1、质子极性溶剂
1)分子结构:含有O-H、N-H等、既是氢键的给予体,又是 氢键的受体
2)作用原理:
通过氢键缔合,它主要促进键的破裂。可以稳定正离子, 也可稳定负离子。有利于SN1反应;但不利于负离子作为亲 核试剂 的SN2。
2、非质子极性溶剂
1)分子结构:不含质子,但存在孤对电子。只是氢键受 体,不是氢键的给予体。
CH3
HO C H + I
C2H5
(S)-2-丁醇
说明:氢氧根离子从 背后进攻、协同反应
六、化学热力学
通过反应中
H 、 S 、 G 的变化来求得反应机理
的信息。
- H = 键 能 生 成 - 键 能 断 裂
负值:放热反应, 正值:吸热反应 放热反应比吸热反应易进行
G = H-T S
自由能值越大,K值就越小,说明对反应不利。
反应) 4、可能中间体的合成
a) 分离出中间体
例如,霍夫曼重排反应中至少有三种中间体是很容易分 离得到的,即N-溴代酰胺及其负离子和异氰酸酯.
分离得到了RCONHBr, 证明了反应确实经过这个中间体.
RCONH2 NaOBr RCONHBr
O
RCN
R-N=C=O H2O
RCONBr -Br
O
RNHC
Ka
[H 3O ][ A ] [HA]
*pka= –lgKa :值越小酸性强;否则相反。
2、碱强度表示法 RNH2+ H2O Kb RNH3+ + OH-
*pKb:值越小,碱性强。它的共轭酸酸性就弱; * pka:RN+H3:值越大,是强碱。
3、酸碱反应进行方向的预测
强碱 A + 强酸 B
NO2+ + H3O+ + 2HSO4 NO2+ + NO3 + H2O
三、催化作用:催化机理与催化剂的作用相符
OR-C-CN
CNR-CHO + HCN
H+
OH RC H
H+
R-CH-CN
CN-
OH
结论:加碱催化使反应加速:说明反应机理是亲核加成
四、同位素标记:
14C、18O 确定反应原子的去向和键断裂位置
* Cl NaNH2 H
* NH3
* NH2
* NH2
*
+
~50%
~50% NH2
说明:机理是先形成苯炔,然后再发生与氨加成
五、立体化学 根据化合物的构型变化可以推断反应物变化的方式,键断裂和 形成的方向。 2-碘丁烷的碱性水解反应:现象构型转化。
CH3
HO + C I
H C2H5
(R)-2-碘丁醇
熵值大小:提供反应类型的信息:
分解反应 消除反应
S 8~17 S -3~4
重排反应 结合反应
S -20~0 S -20~-5
七、化学动力学
可以获得在控制速率步骤中,哪些分子和有多少分子参与 了反应的信息。
例:实验发现1o、2o卤代烃碱性水解时,二级反应,由此可
推出它是两分子亲核取代反应;而3o卤代烃水解时,反应速度与
非O-H、C-H、 N-H键断裂的反 应时
当分子中氢被重氢置换 时,反应速率往往发生 较大改变。
kH / kD= 1 -8
当分子中氢被重氢置换时,反 应速率
kH / kD = 0 -1.5
1、重氢同位素效应
2、次级同位素效应
重氢同位素值越大,说明关键步骤中有X-H键的断裂
例1:丙酮-H的溴代反应历程
二、介电常数与溶剂效应
1、介电常数大小的物理意义: 表示溶剂极性大小,介电常数越大,极性也越大。 >15为极性溶剂; <15为非极性溶剂.
环已烷 2.01 ;水78.55 ;乙酐20.7 ;DMF 36.7 是衡量溶剂隔离正负离子的能力,即:溶剂稳定离子的能力。
正、负离子在介电常数大的溶剂中静电引力小,有利于电离 和分开。
C H
+ N
C
H
H CH
O-
H
S + H
HC
C
HH H
H
练习:
1、比较下列碱性基团的碱性大小
Cl- 、CH3COO- 、OH2、机理分析:同位素示踪在反应机理的研究中起到十分
重要的作用,请给出以下反应的机理
O
H3C
CH3
H3O18
O18
H3C
CH3
3、根据酸碱反应的规律,判断下列反应能否进行?
可能的历程是: 1)控制速率步骤中是溴直接进攻-H