基于信息熵和残基电荷性的酪氨酸硝基化位点预测
蛋白质结构预测
序列基序识别 二硫键识别 折叠子识别 残基接触预测 结构域预测
结构表面识别
预测蛋白质表面结构功能关键区域
5
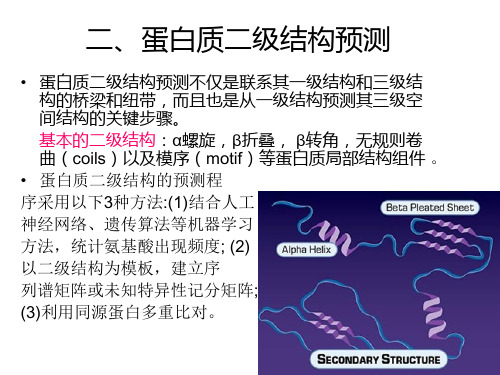
PredictProtein Secondary Structure
PredictProtein Secondary Structure
H:螺旋 E:折叠 L:环 e:暴露表面﹥16%残基 b:其它残基
3
PredictProtein提交界面
序列提交窗口
分析方法程序详解
PROFsec(默认) PROFacc(默认) 序列预测
基于轮廓(profile)的神经网络算法预测蛋 白质二级结构 基于轮廓(profile)的神经网络算法预测残 基溶剂可及性
PHDhtm(默认)
ASP(默认) COILS(默认) PROFtmb ProSite(默认) SEG(默认) PredictNLS(默认) DISULFIND(默认) AGAPE PROFcon ProDom(默认) CHOP ConSeq
22
SWISS-MODEL
• SWISS-MODEL是一个蛋白质3D结构数据库,库中收录的蛋白质结
构都是使用SWISS-MODEL同源建模方法得来的。
– /
• 基于同源建模法与PDB数据库已知结构的蛋白质序列比对 进行预测
23
SWISS-MODEL
蛋白质三维结构预测
方法 特点 工具
同源建模法 基于序列同源比对,对于序列相似度>30% SWISS-MODEL, CPHmodels ( Homology/Comparativ 的序列模拟比较有效,最常用的方法 e modelling ) 线串法/折叠识别法 (Threading/Fold recognition) 从头预测法 ( Ab initio/De novo methods ) “串”入已知的各种蛋白质折叠骨架内,适 于对蛋白质核心结构进行预测,计算量大 基于分子动力学,寻找能量最低的构象, 计算量大,只能做小分子预测
蛋白质修饰位点预测

蛋白质修饰位点预测
蛋白质修饰位点预测是生物信息学领域的一个重要研究方向。
蛋白质修饰是一种在蛋白质翻译后发生的化学变化,对蛋白质的功能和活性产生重要影响。
目前,许多生物信息学方法已经被开发用于预测蛋白质修饰位点,主要包括以下几种:
1. 基于机器学习的方法:这类方法通过训练一个分类器(如支持向量机(SVM)、神经网络等)来预测蛋白质修饰位点。
这类方法通常需要大量的已知修饰位点和非修饰位点的蛋白质序列作为训练数据。
例如,研究人员针对水稻蛋白质磷酸化位点开发了一种基于SVM的预测工具[1]。
2. 基于氨基酸序列特征的方法:这类方法通过分析蛋白质序列中的氨基酸特征(如氨基酸频率、组成等)来预测修饰位点。
这类方法不需要依赖蛋白质结构信息,仅通过序列信息进行预测。
例如,研究人员利用氨基酸频率计算方法来进行特征提取,并结合SVM算法构建了一种针对水稻蛋白质磷酸化位点的预测工具[2]。
3. 基于结构的方法:这类方法通过分析蛋白质三维结构来预测修饰位点。
由于蛋白质结构与功能密切相关,这类方法具有较高的预测准确性。
然而,结构信息通常不易获取,且计算成本较高。
4. 集成学习方法:这类方法将多个预测模型进行集成,以提高预测准确性。
例如,研究人员将多个基于机器学习的预测模型进行集成,构建了一种针对蛋白质翻译后修饰位点的预测工具[3]。
总之,蛋白质修饰位点预测是一个具有挑战性的课题。
随着生物信息学技术的发展,未来可能会出现更多高效、准确的预测方法。
同时,蛋白质修饰位点预测在生物学研究中的应用也将越来越广泛,有助于揭示蛋白质功能和调控机制。
蛋白催化位点预测

蛋白催化位点预测蛋白催化位点预测是一项重要的研究领域,在药物设计和生物工程等领域具有广泛的应用前景。
通过预测蛋白质分子中的催化位点,我们可以更好地理解蛋白质的功能和作用机制,从而为新药的开发和治疗疾病提供重要的指导。
催化位点是蛋白质分子中具有催化活性的特殊位置。
它们通常由氨基酸残基组成,具有特定的结构和功能。
在蛋白质的结构中,催化位点通常位于活性中心附近,与底物分子发生特定的相互作用,从而催化化学反应的进行。
为了预测蛋白质中的催化位点,研究人员通常使用一系列的计算方法和算法。
这些方法可以分为结构基于方法和序列基于方法两大类。
结构基于方法主要是基于蛋白质的三维结构进行预测。
通过分析蛋白质的结构特征,如氨基酸的侧链构象、残基间的相互作用等,可以预测催化位点的位置和性质。
这些方法通常需要蛋白质的结构信息,因此需要进行蛋白质结构的解析和模拟。
序列基于方法则是基于蛋白质的氨基酸序列信息进行预测。
通过分析蛋白质序列中的保守位点和保守模体,可以预测催化位点的位置和特征。
这些方法通常不需要蛋白质的结构信息,因此可以应用于未知结构的蛋白质。
除了结构和序列信息,催化位点的预测还可以利用生物信息学数据库和机器学习算法。
通过分析已知催化位点的特征和模式,可以训练模型来预测未知蛋白质中的催化位点。
这些方法在大规模的蛋白质数据分析中具有较高的准确性和效率。
尽管蛋白质催化位点预测在理论和方法上取得了一定的进展,但仍然存在一些挑战和难题。
首先,蛋白质的结构和功能非常复杂,催化位点的预测仍然存在一定的误差和不确定性。
其次,蛋白质的结构和序列信息在不同的物种和组织中可能存在差异,这也给催化位点的预测带来了一定的困难。
为了进一步提高蛋白质催化位点预测的准确性和效率,需要不断开展基础研究和方法改进。
同时,加强数据共享和合作,建立更加完善和准确的数据库,也是推动该领域发展的重要方向。
蛋白质催化位点预测是一项具有挑战性和重要性的研究课题。
蛋白修饰分析流程

蛋⽩修饰分析流程#流程⼤放送#蛋⽩质修饰分析流程1.背景和意义由mRNA表达产⽣的蛋⽩质需要经过蛋⽩质翻译后的化学修饰,即蛋⽩质修饰,来完成蛋⽩质的特定功能。
化学修饰会引起蛋⽩质的结构和理化改变,进⽽引起蛋⽩质的活性和功能改变。
蛋⽩质修饰包括磷酸化、⼄酰化、糖基化等,是调节蛋⽩质功能的重要⽅式。
例如,蛋⽩质的磷酸化与细胞信号传导、细胞周期调节、⽣长发育以及癌症机理等诸多⽣物学问题具有密切关系;蛋⽩质的⼄酰化是调节蛋⽩质活性的⼀种重要⽅式;蛋⽩质的糖基化对蛋⽩质的三维结构和功能具有重要影响;蛋⽩质的棕榈化对于跨膜蛋⽩质的活性具有重要的调节作⽤;蛋⽩质的硝基化和亚硝基化在蛋⽩质的氧化损伤⽅⾯具有重要作⽤。
因此,对蛋⽩质修饰进⾏详细分析对阐明蛋⽩质的功能具有重要意义。
2. 使⽤范围:细胞信号传导、细胞周期调节、⽣长发育,氧化机制研究,肿瘤与癌症机理研究等。
3.分析步骤:1.找到特定蛋⽩对应的序列⽂件根据蛋⽩质的名称,找到蛋⽩质组中的特定蛋⽩对应的序列⽂件,下载后进⾏后续数据处理,序列下载可⽤⽹站包括NCBI、EBI等。
2.预测蛋⽩质的磷酸化位点、⼄酰化位点和糖基化位点对蛋⽩质组学得到的重要蛋⽩质,进⾏功能位点预测,找到其中可能的磷酸化、⼄酰化、糖基化位点。
磷酸化位点预测对应的软件包括GPS、PhosphoSitePlus等,⼄酰化位点预测对应的软件包括ASEB、NetAcet等,糖基化位点预测对应的软件包括NetNGlyc、DictyOGlyc等。
图1. 预测得到的蛋⽩质磷酸化位点,包含磷酸化的位置、长度和打分信息。
图中红⾊标识的残基为特定蛋⽩中具有磷酸化作⽤的残基位点。
图2. 对于特定蛋⽩预测得到的蛋⽩质N末端的⼄酰化位点,包含⼄酰化的残基名称和打分信息。
图3. 对特定蛋⽩质预测得到的糖基化位点,包含蛋⽩质各个位置的糖基化可能性以及阈值。
3.预测蛋⽩质的棕榈酰化位点、硝基化位点和亚硝基化位点对蛋⽩质进⾏功能位点预测,找到其中可能的棕榈化、硝基化、亚硝基化位点。
氨基酸泛素化位点预测

氨基酸泛素化位点预测
氨基酸泛素化位点预测是一种重要的生物信息学分析方法,它可以帮助科学家们更准确地了解蛋白质的功能和调控机制。
泛素化是一种重要的蛋白质修饰方式,通过在特定氨基酸残基上加上泛素分子,从而改变蛋白质的性质和功能。
因此,预测泛素化位点对于理解蛋白质的功能和调控机制具有重要意义。
在氨基酸泛素化位点预测中,科学家们通常采用基于计算的方法,通过分析蛋白质的序列和结构特征来预测泛素化位点。
其中,一些常见的预测方法包括基于机器学习的预测模型、基于规则的预测方法和基于结构的预测方法等。
基于机器学习的预测模型通常需要使用大量的实验数据来训练模型,并通过模型的学习来预测新的泛素化位点。
这种方法具有较高的预测精度和可靠性,但需要大量的实验数据来支持。
基于规则的预测方法则是通过分析蛋白质的序列特征来预测泛素化位点。
这种方法通常基于一些已知的规则和经验,例如某些氨基酸残基更容易发生泛素化等。
这种方法简单易行,但可能存在一定的误差和局限性。
基于结构的预测方法则是通过分析蛋白质的三维结构来预测泛素化位点。
这种方法需要考虑蛋白质的空间构象和相互作用等因素,因此预测精度较高,但需要更多的计算资源和时间。
总之,氨基酸泛素化位点预测是一项重要的生物信息学分析方法,可以帮助科学家们更深入地了解蛋白质的功能和调控机制。
随着技术的不断发展和完善,相信未来会有更多的预测方法和工具出现,为蛋白质研究带来更多的便利和突破。
thermotogamartima嗜热木聚糖酶化学修饰与其结构特性关系

Thermotoga martima嗜热木聚糖酶化学修饰与其结构特性关系苏樨州,蔡萍,严明*(南京工业大学制药与生命科学学院,材料化学工程国家重点实验室,江苏南京 210009)摘要:应用化学修饰的实验方法,结合蛋白质结构信息的计算来研究酶蛋白中氨基酸残基化学修饰与结构信息之间的关系。
以Thermotoga maritima嗜热木聚糖酶为对象,采用PDB数据库中的1VBR为模板计算其序列中色氨酸、谷氨酸、天冬氨酸的溶剂可及性、氢键、盐桥数等结构特性,并与该酶化学修饰的实验结果相对比。
结果表明酶活性中心3个色氨酸中,可及性大的Trp802与Trp602两个残基对酶的活性影响较大;序列中谷氨酸与天冬氨酸的氢键、盐桥数较多,修饰其对酶的热稳定性有很大影响。
此结果有助于深入了解蛋白质中与化学修饰有关的结构特性,并为基于蛋白质结构的酶蛋白改性奠定了基础。
关键词:嗜热木聚糖酶;化学修饰;结构特性Relationship between structural characteristics and chemical modification to Thermo-stable Xylanase from Thermotoga maritimaSU Xizhou, CAI Ping, YAN Ming(State Key Laboratory of Materials-Oriented Chemical Engineering, College of Life Science and Pharmaceutical Engineering, Nanjing University of Technology, Nanjing 210009, Jiangsu, China)Abstract: Chemical modification and protein structure calculation methods were used to investigate the relationship between the chemical modification of amino-acid residues and their structure informations in protein. Choose thermo-stable xylanase from Thermotoga maritima as research object and 1VBR from PDB as template, computing the structural characteristics of 1VBR such as accessibility, hydrogen bonding network, and salt bridges. Then compare these structural characteristics with experimental data of chemical modification to xylanase. Results show that two tryptophans,Trp802 and Trp602, which near the active site of xylanase are essential for enzyme activity, as they have higher accessibility; glutamates and aspartates have more hydrogen bonding network and salt bridges in the structure, so they are important to the thermal stability of xylanase. These results were helpful for farther study on the structural characteristics of protein which have relationship with their chemical modification, also provide references for protein reshaping based on protein structure.Keywords:thermo-stable xylanase; chemical modification; structural characteristics引言木聚糖酶(EC 3.2.1.8;1,4-b-D-endoxylanase)是木聚糖降解酶系中最关键的酶,在食品、饲料、纺织、造纸工业等方面都有重要的应用价值。
基于免疫信息学的SARS-CoV-2表位筛选及疫苗分子设计分析

基于免疫信息学的SARS-CoV-2表位筛选及疫苗分子设计分析①邓辉雄王革非李雁嫘宋鑫利谷李铭李蕊(汕头大学医学院微生物学与免疫学教研室,病原与免疫学研究中心,汕头515041)中图分类号R392.1文献标志码A文章编号1000-484X(2022)07-0838-10[摘要]目的:基于新型冠状病毒(SARS-CoV-2)全病毒或Spike蛋白作为免疫原可能存在的无关抗原表位干扰,筛选鉴定优势保护性抗原表位,构建候选多价表位疫苗。
方法:以免疫信息学确定诱导主要中和抗体、激活细胞免疫应答的细胞保护性抗原表位,以Raptor X、trRosetta预测蛋白疫苗的二级、三级结构,并对候选蛋白疫苗进行二硫键的设计及对接分析,以C-ImmSim服务器模拟预测分析多表位疫苗的免疫原性和免疫应答。
结果:实验筛选了Spike蛋白免疫原性强的B细胞表位,具有高保守性和人群覆盖度广的IFN-γ、IL-4阳性的Th表位及CTL表位,计算模拟验证了疫苗的免疫应答特性。
结论:信息学分析结果初步显示候选表位疫苗具有良好的平衡体液免疫和细胞免疫应答能力。
[关键词]新冠病毒;Spike蛋白;抗原表位;平衡免疫;免疫信息学Epitopes screening and vaccine molecular design of SARS-COV-2based on immunoinformaticsDENG Huixiong,WANG Gefei,LI Yanlei,SONG Xinli,GU Liming,LI Rui.Department of Microbiology and Immu⁃nology,Pathogen and Immunology Research Center,Shantou University Medical College,Shantou515041,China [Abstract]Objective:Based on SARS-CoV-2whole virus or Spike protein as an immunogen may be interference with the epitopes of unrelated antigens.Constructed of candidate vaccine by screening and identifying the dominant protective epitopes.Methods:Immunoinformatics was used to identify the protective epitopes that induce the main neutralizing antibodies and activate the cellular immune response,Raptor X and trRosetta were used to predict the secondary and tertiary structures of the vaccine,and disulfide bond design and docking analysis were carried out for candidate protein vaccine,immunogenicity and immune response of multi-epitopes vaccine were analyzed using C-ImmSim server.Results:B cell epitopes on strong immunogenicity of Spike protein,highly conservative and wide coverage of population and positive IFN-γ,IL-4of Th epitopes and CTL epitopes were selected,and the vaccine immune response characteristics were verified in silico.Conclusion:Results of immuneinformatics analysis preliminarily show that the candi‐date vaccine had a well-balanced on humoral and cellular immune response.[Key words]SARS-CoV-2;Spike protein;Epitopes;Immunebalance;Immunoinformatics新型冠状病毒(SARS-CoV-2)感染引起的新冠肺炎(Corona Virus Disease2019,COVID-19)疫情在全球200多个国家和地区流行,对全球公共卫生安全造成巨大威胁,给社会生活及经济发展造成巨大损失。
模式识别与生物信息学在蛋白质相互作用预测中的应用研究

模式识别与生物信息学在蛋白质相互作用预测中的应用研究蛋白质相互作用是生命体内复杂的分子交互作用过程之一,对于揭示细胞内信号传导、代谢途径以及疾病发生机制具有重要意义。
在过去的几十年中,科学家们不断探索蛋白质相互作用的研究方法,其中模式识别和生物信息学技术因其高效且准确地预测蛋白质相互作用而受到广泛关注。
模式识别是一种通过从已知数据中提取特征并通过这些特征来识别新数据的技术。
在蛋白质相互作用预测中,模式识别技术可以通过学习训练集数据中的模式或特征,并使用这些模式来预测新蛋白质相互作用。
这些模式可以是某些氨基酸残基在蛋白质结构中的位置和性质,也可以是蛋白质序列或结构的一些统计特征。
通过使用机器学习算法,如支持向量机(SVM)和随机森林(Random Forest),模式识别可以对蛋白质相互作用进行分类和预测。
生物信息学是一门综合了生物学、计算机科学和统计学的学科,其研究的重点是在生物学中收集、存储、管理和分析大规模的生物数据。
在蛋白质相互作用预测中,生物信息学技术可以从多个维度获取蛋白质的信息,并将其应用于模型训练和预测。
例如,通过分析蛋白质序列和结构的一些特征,如氨基酸组成、疏水性和电荷等,并将其编码为数学描述,可以构建有助于蛋白质相互作用预测的模型。
在蛋白质相互作用预测领域,模式识别和生物信息学技术的应用已取得一系列显著成果。
一些研究表明,通过结合多种特征和算法,可以提高蛋白质相互作用预测的准确性。
例如,研究人员可以结合蛋白质序列、结构、功能域和进化信息等多个特征进行预测,并使用机器学习算法对这些特征进行集成。
此外,一些研究还将互作网络中的拓扑结构信息与模式识别和生物信息学技术相结合,以进一步提高预测准确性。
尽管模式识别和生物信息学技术在蛋白质相互作用预测中取得了显著进展,但仍面临一些挑战。
首先,蛋白质相互作用是一种复杂的现象,受许多因素的影响,如结构适配性、动力学和环境条件等。
因此,单一的模式识别和生物信息学技术可能无法完全捕捉到这种复杂性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
中 图分 类 号 : 8 1 T 3 1 Q 1 ; P 9 文献标志码 : A
e . n。 du e
南 昌 大学 学报 ( 科 版 ) 理
生 物半 衰期 、 免疫应 答 和 酪 氨酸 磷 酸 化 调节 的信 号
转 导过 程盟 。 已 经 证 实 , 括 糖 尿 病 心 血 管 并 发 ] 包 症_、 3 缺血 一再 灌 注损 伤 和 阿 兹海 默 症 等 各 种 神 ]
c n n l e e y o i e nir to a t i fu nc son t r s n t a i n.
Ke r s Ty osne n t a i y wo d : r i ir ton;nf m a in nt o i or to e r py; ha g fr sd c r e o e i ue
b De a t n fM a h ma is Na c a g Un v r i Na c a g 3 0 3 , ia; . p rme to t e t , n h n i e st c y, n h n 3 0 1 Ch n
2. pa t e fSce c nd A r s Na ha g M iia y A c e y, De rm nto in e a t , nc n lt r ad m Nan ha g 3 01 c n 3 03, i a) Ch n
Absr c A w e ho as d o nf r to e r py a ha g f r s d s de l pe o p e i tt ta t: ne m t d b e n i o ma i n nt o nd c r e o e i ue wa veo d t r d c he t o i ir ton st s By usn h 一ol r s v ld to t e p e i tvea c r c nd M a t ws Cor ea yr sne n t a i ie . i g t e 1 f d c os — a i a i n, h r d c i c u a y a the 0 r l- ton Co fii nto he m o lwe e 84 0 i e fce ft de r .8 a d 6 .6 , e p c i e y n 9 9 r s e tv l .So e i i r s u so r me pr l na y dic s i ns we e m
第 3 卷 第 3期 6
21 0 2年 6月
南 昌 大 学 学报 ( 科 版 ) 理
J u n l fNa c a g U nv r i ( t r l ce c ) o r a o n h n i e st Na u a in e y S
Vo - 6 NO 3 l3 .
wh c v r o e h o t a it n t a h h r e t e s q e c se s o l s n o ma i n a d t er — ih o e c m d t ec n r d c i h tt e s o tp p i e u n e wa a y t o e i f r t n h e o d o
蛋 白质序 列 收集 自科 学 文 献 ( 过 搜 索 关 键 词 “ i 通 n—
tain 或 “ i ae ” 和 d P M[] S s T [ 以 rt ” nt t理 , 发 现药 物 作 用靶 点 提 供 为
化 有 2种主要 途径 : 一种 是 含 卟 啉过 氧化 物 酶介 导
的 , 一 种 是 通 过 活 性 氮 (rat ent g n s e 另 eci i o e p — v r
ce , NS 如 过 氧 亚 硝 酸 根 离 子 ( eo y i i , i R ) s p rx nt t re
新 思路 。
及 UnP of ir t 公 共数 据库 ;i 为 了避 免预测 结果 过 1 i ) 拟合 , 使用 C - T[ D HI 】 设定 序列 一致 性 阈值 4 对 0
蛋 白序列 进行 聚类 ;I 采 用滑 动窗 口策略提 取含 有 i) i
目前 , 鉴定 蛋 白质 翻 译 后修 饰 位点 的实验 方 法 主要 有 特 异 性 抗 体 、 片 技 术 和 质 谱 分 析[ 等 。 芯 1 然 而 , 用实 验方 法鉴 定 翻译 后修 饰 位 点 的条 件 要 使 求高 , 费时、 且 费力 和 费用 昂贵 。因此 , 展计 算 预 发 测 方法识 别 蛋 白质 翻译后 修饰 位点成 为 近年来 的研
me to y o i er sd e ,h d e te ou in rl o s r e ie n o g r n estsh d s mesg ii n ft r sn e iu s t ea jc n v l t a i c n e v d stsa d l n —a g ie a o i nf- a o y
d n a ti fr t nwo l eito u e yj s n r a ig t eln t fp p ie Th rd cin p ro m— u d n n o mai ud b n r d c db u tic e sn h e g h o e td . ep e ito ef r o
a c ft e mo e s u tma ey i r v d F a u e a a y i r v a e h t t e l c le e t o t tc e v r n n e o h d l wa li t l mp o e . e t r n l ss e e ld t a h o a l c r s a i n i - o
Pr d c i n o y o i e n t a i n s t s b s d o nf r a i n e i to f t r sn ir to ie a e n i o m to e t o nd c r e o e i u n r py a ha g f r s d e
ma e t hewi o o nf ma i n e r y a r d ton lc tnu uswi o Ou e u t h d o t nd w fi or to ntop nd t a ii a on i o nd w. rr s ls s owe ha he d t tt wi o o n o m a i n e r py c l fe tvey c p u e t mpo t n ie n t e nir — y osn e i nd w fi f r to nto ou d e f c i l a t r he i r a t st s i h to t r i e p ptde,
蛋 白质硝基 化是 一种 与氧化 应激 密切 相关 的可 逆 的蛋 白质 翻 译 后 修 饰 ( rti p s— a sain l p oe ott n l o a n r t mo ict n P df ai , TM) 它通 常是指 硝基结 合 到酪氨 酸 i o , 残基 芳环 上生成 3 一硝 基 酪 氨酸 ( 一ntoyo ie 3 i tr s , r n
1 实验 数 据 与 方法
1 1 数 据 集 .
经 退行 性 疾病 在 内的 8 ] O多种 疾 病 中均 伴 随着 酪 氨 酸硝基 化水 平 的升 高 , 表 明酪 氨 酸 残基 的硝 基 这 化 可能参 与疾 病 的发 生和 发展 过程 。酪氨 酸硝基 化
蛋 白的鉴 定将 极大 地 促 进结 构 分 析 , 高对 特 定 酪 提 氨 酸 残基 直 接硝 基 化原 因 的理解 , 而 有助 于揭 ]从
J n 2 1 u.02
文 章 编 号 : 0 60 6 ( 0 2 0 — 2 50 1 0 —4 4 2 1 ) 30 4 — 5
基 于 信 息 熵 和 残 基 电荷 性 的 酪 氨 酸 硝 基 化 位 点 预 测
施 绍 萍 h , 志 勇 邱 丁h , 揭 , 建
(. 昌 大 学 a 化 学 系 , 西 南 昌 3 0 3 ;. 学 系, 西 南 昌 30 3 ; 1南 . 江 3 0 1b 数 江 3 0 l 2 南 昌 陆 军 学 院科 文教 研 室 , 西 南 昌 3 00 ) . 江 3 1 3
本文 所 用 训 练 数 据 集 包 含 5 4条 蛋 白 质 , 5 由
L u等口]0 0年构建 , 中经 实 验验 证 的 非冗 余 蛋 i 。 1 2 其
白质酪氨 酸硝基 化位 点有 1 6 个 , 06 酪氨酸非 硝基 化 位 点 78 6 4个 。该数 据 集 的具 体 构建 过 程 如下 :i )
摘
要 : 用 氨 基 酸 残基 信息 熵 优 化 窗 口, 合 氨 基 酸 的 电 荷 性 构 建 了蛋 白 质 酪 氨 酸 硝 基 化 位 点 的 预 测 模 型 。基 采 结
于 1 一倍 交 叉 验 证 , 模 型 的预 测 准 确 率 和 马 氏相 关 系 数 分 别 达 到 了 8. O 和 6 . 9 。 同时 , 信 息 熵 优 化 窗 O 该 4 8 9 6 对
口和 传 统 连 续 窗 口进 行 了 探 讨 , 果 表 明 , 息 熵 窗 口能 够有 效捕 获 酪 氨 酸 硝基 化 肽 段 上 的 重要 位 点 , 服 短 肽 序 结 信 克
列 易 丢 失 信 息 而 单 纯 增 大 肽 段 长 度 又 会 引 入 冗 余 信 息 的 矛 盾 , 最 终 提 高 模 型 的 预 测 性 能 。特 征 分 析 揭 示 酪 氨 酸 并
S h o pn , I h— o g , U in d n HIS a — i g JE Z i n 。 QI Ja — ig y