FluorCam叶绿素荧光成像一
Fluorcam多光谱荧光成像技术及其应用

FluorCam多光谱荧光成像技术(Multi-color FluorCam)自上世纪90s年代PSI公司首席科学家Nedbal教授与公司总裁Trtilek博士等首次将PAM脉冲调制叶绿素荧光技术与CCD技术结合在一起,成功研制生产FluorCam叶绿素荧光成像系统(Nedbal等,2000)以来,FluorCam叶绿素荧光成像技术得到长足发展和广泛应用,先后有封闭式、开放式(包括标准版和大型版)、便携式叶绿素荧光成像系统,及显微叶绿素荧光成像系统、大型叶绿素荧光成像平台(包括移动式、样带式、XYZ三维扫描式等)等,近些年还进一步发展了PlantScreen植物表型成像分析平台(Phenotyping)(有传送带版、XYZ三维扫描版及野外版等)及多光谱荧光成像技术。
Multi-color FluorCam多光谱荧光成像技术包括多激发光-多光谱荧光成像技术和UV 紫外光激发多光谱荧光成像技术:1.多激发光-多光谱荧光成像技术:通过光学滤波器技术,仅使特定波长的光(激发光)到达样品以激发荧光,同时仅使特定波长的激发荧光到达检测器。
不同的荧光发色团(如叶绿素或GFP绿色荧光蛋白等)对不同波长的激发光“敏感”并吸收后激发出不同波长的荧光,根据此原理可以选配2个或2个以上的激发光源、绿波轮及相应滤波器,对不同波长荧光(多光谱荧光)进行成像分析。
如FluorCam便携式GFP/Chl.荧光成像仪及FluorCam封闭式GFP/Chl.荧光成像系统具备红光和兰光及相应滤波器,可以对GFP和叶绿素荧光成像分析;FluorCam开放式多光谱荧光成像系统可以进一步选配不同颜色的激发光,如除红光、蓝光外,还可选配绿色光源及相应滤波器,以对YFP进行荧光成像分析等;2.UV紫外光激发多光谱荧光成像技术:长波段UV紫外光(320nm-400nm)对植物叶片激发,可以产生具有4个特征性波峰的荧光光谱,4个波峰的波长为兰光440nm(F440)、绿光520nm(F520)、红光690nm(F690)和远红外740nm(F740),其中F440和F520统称为BGF,由表皮及叶肉细胞壁和叶脉发出,F690和F740为叶绿素荧光Chl-F。
植物表型组学研究技术(一)FluorCam 叶绿素荧光成像技术
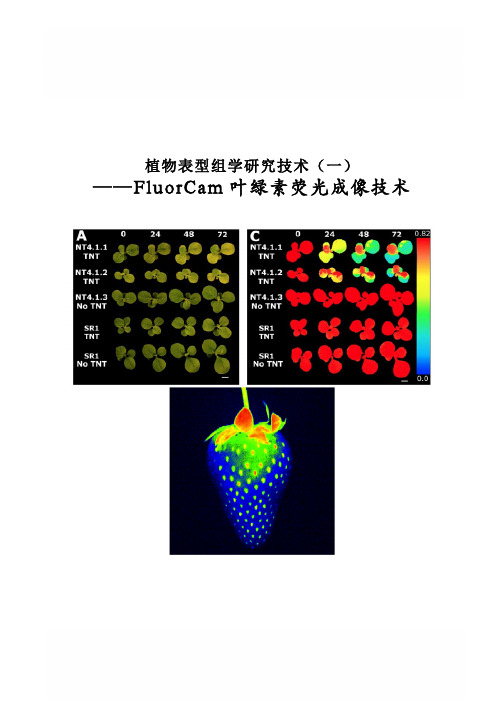
植物表型组学研究技术(一) ——FluorCam叶绿素荧光成像技术FluorCam叶绿素荧光成像技术Rousseau等(High throughput quantitative phenotyping of plant resistance using chlorophyll fluorescence image analysis.Plant Methods, 2013, 9:17),利用FluorCam开放式叶绿素荧光成像系统作为高通量表型分析平台,采用图像阈值分割等分析方法,对植物病原体感染进行了定量分析检测,根据Fv/Fm将感染分为不同阶段/等级,特别是可以将用其它方法难以分辨出来的感染前期加以分辨,并对5个品种的菜豆对普通细菌性疫病的抗性进行了定量分析评价。
PSI公司首席科学家Nedbal教授与公司总裁Trtilek博士等首次将PAM叶绿素荧光技术(Pulse Amplitude Modulated technique——脉冲调制技术)与CCD技术结合在一起,于1996年在世界上成功研制生产出FluorCam叶绿素荧光成像系统(Heck等,1999;Nedbal等,2000;Govindjee and Nedbal, 2000)。
FluorCam叶绿素荧光成像技术成为上世纪90年代叶绿素荧光技术的重要突破,使科学家对光合作用与叶绿素荧光的研究一下子进入二维世界和显微世界,广泛应用于植物生理生态、植物胁迫与抗性监测、作物育种、植物表型分析等。
不同于其它成像分析技术,FluorCam叶绿素荧光成像只对叶绿素荧光波段敏感,可以有效避免环境光的干扰,特异性、高灵敏度反映植物生理生态状况。
主要功能特点如下:1)高灵敏度CCD,时间分辨率可达50帧/秒,有效抓取叶绿素荧光瞬变;可选配高分辨率CCD,分辨率1392x1040像素,用于气孔功能成像分析、稳态荧光如GFP荧光测量等2)具备完备的自动测量程序(protocol),可自由对自动测量程序进行编辑:a)Fv/Fm:测量参数包括Fo,Fm,Fv,QY等b)Kautsky诱导效应:Fo,Fp,Fv,Ft_Lss,QY,Rfd等荧光参数c)荧光淬灭分析:Fo,,Fm,Fp,Fs,Fv,QY,ΦIINPQ,Qp,Rfd,qL等50多个参数d)光响应曲线LC:Fo,Fm,QY,QY_Ln等荧光参数e)PAR吸收f)GFP等静态荧光测量g)OJIP与JIP-test(FKM与封闭式荧光成像系统):Fo,Fj,Fi,P 或Fm,Mo(OJIP曲线初始斜率)、OJIP固定面积、Sm(对关闭所有光反应中心所需能量的量度)、QY、PI等26个参数3)自动重复实验功能,可无人值守自动循环完成选定的实验程序,重复次数及间隔时间客户自定义,成像测量数据自动按时间日期存入计算机4)FluorCam成像分析软件:具在线功能(Live)、实验程序选配功能(Protocols)、成像预处理功能(Pre-processing)及成像分析结果展示报告功能(Result)四大功能模块a)在线功能(live):可对仪器和样品进行在线测试调试、快照、显示实验进度、在线显示荧光瞬变动态视频等b)实验程序选配功能(protocols):可选配不同的实验程序,并可对实验程序进行编辑、设置、储存(以备以后使用同样的实验程序)等c)成像预处理功能:可浏览整个测量视频及任何点、任何区域的荧光动态变化曲线,可进行“选区操作”或“分级操作”(图像阈值分割功能);选区操作可对成像进行自动或手动选区(ROI),还可使用“模具”包括多孔板模具、培养皿模具、桌面模具进行模具选区;分级操作具备荧光强度刻度标尺和四个“游标”,通过移动4个游标可以将成像按不同强度划分成不同的荧光范围组进行分析处理,可设置不同的阈值进行图像阈值分割d)结果展示报告功能:可展示所有选区(ROI)的叶绿素荧光参数值及其图像、每个参数的频率直方图及每个ROI的荧光动态图等,可对原数据(kinetic)、叶绿素荧光参数等导出到excel表,还可对每个参数成像图存储成位图5)数据分析具备“信号计算再平均”模式(算数平均值)和“信号平均再计算模式”两种功能模式,在高信噪比的情况下选用“信号计算再平均”模式,在低信噪比的情况下选择“信号平均再计算”模式以过滤掉噪音带来的误差FluorCam叶绿素荧光参数:参数符号概念描述Size 面积(像素值),经校准可测量实际面积Fo 暗适应后的最小荧光Fo_Dn 暗松弛最小荧光,红外光诱导PSIFo_Ln 光适应后的最小荧光,红外光诱导PSIFo_Lss 光适应后稳态最小荧光,红外光诱导PSIFm 暗适应后最大荧光Fm_Dn 暗松弛最大荧光Fm_Ln 光适应最大荧光Fm_Lss 光适应稳态最大荧光Fp Kautsky诱导效应最大荧光Ft_Dn 暗松弛即时荧光Ft_Ln 光适应即时荧光Ft_Lss 光适应稳态荧光Fv Fm-FoNPQ_Dn 暗松弛非光化荧光淬灭,=(Fm-Fm_Dn)/Fm_DnNPQ_Ln 光适应非光化荧光淬灭,=(Fm-Fm_Ln)/Fm_LnNPQ_Lss 稳态非光化荧光淬灭,=(Fm-Fm_Lss)/Fm_LssqP_Dn 暗松弛光化学荧光淬灭,=(Fm_Dn−Ft_Dn)/Fm_Dn−Fo_DnqP_Ln 光适应光化学淬灭,=(Fm_Ln−Ft_Ln)/(Fm_Ln−Fo_Ln)qP_Lss 稳态光适应光化学淬灭,=(Fm_Lss−Ft_Lss)/(Fm_Lss−Fo_Lss)qL_Ln 基于“Lake”模型的光适应光化学淬灭qL_Lss 基于“Lake”模型的稳态光适应光化学淬灭QY_Dn 暗松弛光量子效率,=(Fm_Dn−Ft_Dn)/Fm_DnQY_Ln或ΔF/Fm 光适应光量子效率,=(Fm_Ln−Ft_Ln)/Fm_LnQY_Lss 稳态光量子效率,=(Fm_Lss−Ft_Lss)/Fm_LssFv/Fm或QY_max 最大光量子效率Fv/Fm_Ln 光适应光量子效率,=(Fm_Ln−Fo_Lss)/Fm_LnFv/Fm_Lss 稳态光量子效率,=(Fm_Lss−Fo_Lss)/Fm_LssRfd_Ln 光适应荧光衰减率,用于评估植物活力,=(Fp−Ft_Ln)/Ft_LnRfd_Lss 稳态荧光衰减率,用于评估植物活力,=(Fp−Ft_Lss)/Ft_Lss除上述叶绿素荧光参数外,还可以成像测量PAR吸收、植物光谱反射指数NDVI等,叶片大小(或植物大小)可以反映植物的生长等。
FluorCam叶绿素荧光成像文献 2012 Genetic Analysis of the Hox Hydrogenase

Genetic Analysis of the Hox Hydrogenase in theCyanobacterium Synechocystis sp.PCC 6803Reveals Subunit Roles in Association,Assembly,Maturation,and Function *□SReceived for publication,June 15,2012,and in revised form,November 7,2012Published,JBC Papers in Press,November 8,2012,DOI 10.1074/jbc.M112.392407Carrie Eckert ‡1,Marko Boehm ‡§,Damian Carrieri ‡,Jianping Yu ‡,Alexandra Dubini ‡,Peter J.Nixon §,and Pin-Ching Maness ‡From the ‡Biosciences Center,National Renewable Energy Laboratory,Golden,Colorado 80401and the §Department of Life Sciences,Imperial College London,South Kensington Campus,London SW72AZ,United KingdomHydrogenases are metalloenzymes that catalyze 2H ؉؉2e ؊7H 2.A multisubunit,bidirectional [NiFe]-hydrogenase has been identified and characterized in a number of bacteria,including cyanobacteria,where it is hypothesized to function as an electron valve,balancing reductant in the cell.In cyanobac-teria,this Hox hydrogenase consists of five proteins in two func-tional moieties:a hydrogenase moiety (HoxYH)with homology to heterodimeric [NiFe]-hydrogenases and a diaphorase moiety (HoxEFU)with homology to NuoEFG of respiratory Complex I,linking NAD(P)H 7NAD(P)؉as a source/sink for electrons.Here,we present an extensive study of Hox hydrogenase in the cyanobacterium Synechocystis sp.PCC 6803.We identify the presence of HoxEFUYH,HoxFUYH,HoxEFU,HoxFU,and HoxYH subcomplexes as well as association of the immature,unprocessed large subunit (HoxH)with other Hox subunits and unidentified factors,providing a basis for understanding Hox maturation and assembly.The analysis of mutants containing individual and combined hox gene deletions in a common parental strain reveals apparent alterations in subunit abun-dance and highlights an essential role for HoxF and HoxU in complex/subcomplex association.In addition,analysis of indi-vidual and combined hox mutant phenotypes in a single strain background provides a clear view of the function of each subunit in hydrogenase activity and presents evidence that its physiolog-ical function is more complicated than previously reported,with no outward defects apparent in growth or photosynthesis under various growth conditions.Hydrogenase enzymes are a unique and diverse family of metalloenzymes widely distributed throughout Archaea,Pro-karyotes,and some unicellular Eukaryotes that catalyze the reduction/oxidation of H ϩ/H 2.These enzymes are classified by their metal-containing active sites and include [Fe]-hydroge-nases,[FeFe]-hydrogenases,and [NiFe]-hydrogenases (1).The [NiFe]-hydrogenases are minimally heterodimeric,consisting of a large catalytic subunit and a small subunit containing at least one [FeS]cluster that functions in electron transfer to and from the large subunit (1).The maturation of [NiFe]-hydroge-nases involves at least six maturation proteins (HypABCDEF)essential for the assembly of the [NiFe]active site.Some [NiFe]-hydrogenases additionally require a specialized protease that cleaves the large subunit’s C terminus,a step required for hydrogenase function (2).Research in Escherichia coli suggests that the hydrogenase small subunit only associates with the large subunit after it is fully processed (3).Among the diverse hydrogenases,the bidirectional [NiFe]-hydrogenase (Hox)in cyanobacteria is of great interest for basic biological study as well as for the development of solar hydrogen production technologies (4–6).NAD(P)-linked Hox hydrogenases have been identified and characterized in cyano-bacteria (4,5),the Gram-positive bacterium Rhodococcus opacus (7,8),the Gram-negative bacterium Ralstonia eutropha (6),and the purple sulfur photosynthetic bacteria Thiocapsa roseopersicina and Allochromatium vinosum (9–12).These Hox hydrogenases are multimeric with at least four related sub-units expressed from a single operon.HoxH and HoxY form the hydrogenase moiety and are homologous to the large and small subunits of prototypical heterodimeric [NiFe]-hydrogenases,respectively (1).The [FeS]cluster-containing subunits HoxF,HoxU,and,in some organisms,HoxE form the diaphorase moi-ety (13,14)that catalyzes the oxidation/reduction of NAD(P)H/NAD(P)ϩ(via FMN and NAD binding sites in HoxF)coupled to the hydrogenase moiety (HoxYH)(10,12–15).The R.eutropha Hox hydrogenase does not contain HoxE and instead harbors an unrelated fifth subunit,HoxI,which functions in linkage to NADPH (16).*This work was supported by the National Renewable Energy Laboratory’sLaboratory Directed Research and Development Program (to P.M.,J.Y.,C.E.,and D.C.),the United States Department of Energy Fuel Cell Technol-ogies Program (contract number DE-AC36-08-GO28308)(to P.M.and J.Y.),the United States Department of Energy Biological and Environmental Research Program (contract KP160103)(to M.B.and A.D.),and Engineer-ing and Physical Sciences Research Council Grant (EP/F002070X/1)(to M.B.and P.N.).□SThis article contains supplemental Table 1and Figs.1–5.1To whom correspondence should be addressed:Biosciences Center,NREL,15013Denver West Pkwy.,Golden,CO 80401.Tel.:303-384-6891;Fax:303-384-7836;E-mail:carrie.eckert@.THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL.287,NO.52,pp.43502–43515,December 21,2012Published in the U.S.A.at NATL RENEWABLE ENERGY, on January 16, 2013 Downloaded from /content/suppl/2012/11/08/M112.392407.DC1.htmlSupplemental Material can be found at:Purification of the Hox hydrogenase has been performed in some of the organisms it has been characterized in with varied results.In R.eutropha,the purified Hox complex is Hox-FUYHI2,although HoxI(not present in other characterized Hox hydrogenases)dissociates easily upon more stringent puri-fication conditions(which is why the complex was initially characterized as HoxFUYH)(16).Other purifications in R.eutropha utilizing gene knockouts and ectopic expression demonstrate that HoxYH and HoxFU subcomplexes can be iso-lated,although these subcomplexes were reportedly unstable (13).In A.vinosum(12)and the cyanobacterium Gleocapsa alpicola CALU743(17),attempted purifications of native com-plex resulted in isolation of only HoxYH,whereas in T.roseop-ersicina(18)and Synechocystis(19,20),intact HoxEFUYH complexes were purified.Purification of the Synechocystis Hox hydrogenase by Schmitz et al.(19)resulted in a dimer of a 1:1:1:1:1HoxE/F/U/Y/H complex via a series of column chro-matography steps,whereas Germer et al.(20)purified a com-plex with a0.2:2:2:1:1HoxE/F/U/Y/H ratio using a StrepII-tagged HoxF,raising questions about Hox complex assembly and association in vivo.The physiological role of Hox hydrogenase varies,depending on the organism in which it is present,but it generally functions in anoxic/micro-oxic conditions due to a reversible inactivationby O2(1).In T.roseopersicina,Hox hydrogenase catalyzes H2production under dark,fermentative conditions and in lightwhen thiosulfate is present and alternatively functions in H2 uptake in light when O2is absent(11).The Hox hydrogenase ofR.eutropha(soluble hydrogenase)functions in H2oxidation linked to the regeneration of NADH in support of carbon fixa-tion(6).In cyanobacteria,Hox hydrogenase is expressed under both anaerobic and aerobic conditions(4,21)but is only active under dark,fermentative conditions and in the transition fromdark to light prior to inhibition by O2generated during photo-synthesis(22–24).Respiration and nitrate assimilation mutantsexhibit increased H2photoevolution rates under low O2condi-tions,whereas defects in photosynthetic reactions/ratios have been reported in Hox hydrogenase mutants(15,22–26).There-fore,it is hypothesized that the Hox hydrogenase functions as an electron valve for cells by H2production/oxidation in response to changes in redox states(25,26).HoxEFU are the only homologs for NuoEFG of respiratory complex I in cyano-bacteria,but there is no evidence to date that these diaphorase subunits play any role in respiration(14,27–30).In this work,we generated a series of Hox hydrogenase mutants in a common parental strain,deleting individual hox genes or combinations of hox genes in the unicellular cyanobac-terium Synechocystis sp.PCC6803.This comprehensive collec-tion of hox mutants enabled us to perform systematic studies of mutation effects on complex/subcomplex formation and com-position,subunit abundance,and hydrogenase activity.In addi-tion,we also provide data for growth,photosynthesis,and fer-mentation in a hox operon deletion mutant compared with wild type(WT),revealing that some previously reported hox mutant phenotypes may have been due to differences in strain back-grounds(31,32).EXPERIMENTAL PROCEDURESStrain Background/Construction—All hox mutants weregenerated in a Synechocystis sp.PCC6803glucose-tolerant strain from Teruo Ogawa,who also provided the whole operon deletion,hoxϪ::hygromycin resistance.The hoxHϪdeletion was constructed in a pSMART LC-Amp vector(Lucigen,Inc.) containing a3.6-kb region spanning hoxY and hoxH cut with SspI(removing816bp of hoxH)to insert the kanamycin resist-ance cassette from pUC4K.All remaining hox deletion strains were created by two-step fusion PCR(33)for open reading frame(ORF)replacement,combiningϳ600-bp5Ј-and3Ј-flanking regions for each hox gene ORF replaced with the fol-lowing antibiotic resistance gene ORFs:hoxEϪ::aac3ia(gen-tamicin resistance),hoxFϪ::ermC(erythromycin resistance), hoxUϪ::aph(kanamycin resistance),and hoxYϪ::dfra17(spec-tinomycin resistance).An slr0168neutral site integration vec-tor containing a plastocyanin(petE)promoter for ectopic gene expression(34)was altered by the addition of a225-bp frag-ment from pETDuet containing the T7terminator into the PpuMI restriction site.PCR primers to amplify HoxYH or HoxH from genomic DNA were constructed with sequence encoding His6/HRV3C protease site at the N terminus and5Ј-and3Ј-specific SapI sites for insertion behind the petE pro-moter following restriction digestion and ligation.The pPSBA2KS vector for the integration behind the psbAII pro-moter(35)was altered by removal of a SalI site via partial digest and blunting to allow for retention of the kanamycin resistance gene.PCR primers were designed for amplification and inser-tion of an N-terminal His6-tagged hoxE gene between NdeI and SalI sites.Transformations were conducted by incubatingϳ1g of linear purified fusion PCR products or targeted integra-tion vectors with200l of cells(adjusted to OD730ϭ2.5fromcell cultures at OD730ϭ0.2–0.5)for6h,followed by the addi-tion of2ml of BG11,24-h outgrowth in culture tubes under standard growth conditions,and plating of200l on BG11 plates with antibiotics as required for selection(200g/ml hygromycin or kanamycin,100g/ml gentamicin,erythromy-cin,spectinomycin,or chloramphenicol).Strain Growth—Cultures were inoculated into BG11medium(ATCC medium616)supplemented with3M NiCl2,20m M TES(unbuffered),100m M NaHCO3,and antibiotics as required(one-half concentrations as used on plates listedabove)at an initial OD730ϭ0.05and were grown by shaking inculture flasks with5%CO2under50microeinsteins2(E)mϪ2 sϪ1continuous light from cool white fluorescent bulbs.Cul-tures were generally grown to logarithmic/linear growth phase(OD730ϭ0.2–0.8)for analysis.FPLC Analysis of Synechocystis sp.PCC6803WT Soluble Extract—Soluble extract was prepared from a WT50-ml liquidculture at an OD730Ͼ1(stationary phase).Cells were har-vested,resuspended,and washed twice in ACA buffer(750m M ⑀-amino caproic acid,50m M BisTris/HCl,pH7.0,0.5m M EDTA).Approximately200l of glass beads(150–212m;2The abbreviations used are:E,microeinsteins;MV,methyl viologen;BN,blue native;PSI and PSII,photosystem I and II,respectively;ETR,electron transport rate;BisTris,2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.Cyanobacterial Hox Hydrogenase Complex Association and Functionat NATL RENEWABLE ENERGY, on January 16, Downloaded fromSigma-Aldrich)were added to the cell suspension(ϳ500l), and cells were broken using a Disruptor Genie digital multi-place vortexer(Scientific Industries)at4°C for5min,setting 3,000,followed by centrifugation for10min at maximum speed in a microcentrifuge.Subsequently,2l of DNase I(10,000 units/ml,Thermo Scientific)was added,and the sample was spun again in an ultracentrifuge at100,000ϫg for30min at 4°C.For FPLC,the resulting supernatant was normalized toOD650ϭ10(typically,the OD650of a1:20dilution was mea-sured).To solubilize any remaining thylakoid membrane frag-ments,-dodecyl-D-maltoside was added to a final concentra-tion of0.5%(w/v)from a10%(w/v)stock solution.100l of this soluble extract was loaded onto a Superdex20010/300size exclusion column(GE Healthcare)connected to an Akta Puri-fier FPLC system(GE Healthcare).The sample was run at a flow rate of0.5ml minϪ1with50m M Tris,pH7.5,100m M NaCl,and 0.03%(w/v)-dodecyl-D-maltoside as the buffer.0.5-ml frac-tions were collected by a Frac-950fraction collector(GE Healthcare).A high molecular weight marker mix(GE Health-care)was run as a control for relative protein sizes in the frac-tions(supplemental Fig.1).Protein Preparations,Two-dimensional Blue Native/SDS-PAGE,Affinity Purification,and Western Blotting—Soluble extracts for two-dimensional blue native/SDS-PAGE(BN/ SDS-PAGE)were prepared as described for FPLC analysis above for WT and hox mutant strains,and the extracts werenormalized to an A650ϭ0.6.-Dodecyl-D-maltoside was addedto a final concentration of0.5%(w/v),and the same volume ofCoomassie loading solution(750m M⑀-amino caproic acid,5% (w/v)Coomassie-G)was added to the sample.Subsequently,20l of the sample was loaded per lane and run on a12%(w/v) polyacrylamide BN PAGE first dimension gel.The proteincomplexes in the first dimension gel strips were denatured for1h in solubilization buffer(66m M Na2CO3,2%(w/v)SDS,2%(v/v)-mercaptoethanol,and4M urea)prior to being layered on17.5%(w/v)polyacrylamide,6M urea two-dimensional SDS-polyacrylamide gels(36).The resultant two-dimensional gels were either Coomassie-stained,silver-stained(37),or electro-blotted onto nitrocellulose membranes.For WT and hox mutant one-dimensional Western blots and affinity purifications,cells were resuspended in20m M phos-phate buffer,pH7.4,plus protease inhibitors(Fermentas)and broken by bead disruption as described above.Protein concen-trations were determined by Bradford assay(Fermentas),and levels of each sample were adjusted toϳ100g/ml.For purifi-cation of His6-tagged proteins/complexes,Co2ϩbeads(Pierce)were added to cleared lysates and incubated for1h at4°C,and washes were performed using5times bead volume with20m M phosphate buffer,pH7.4,with or without0.5M NaCl through reloadable columns(Pierce).Beads were resuspended in a50% slurry in buffer.To rule out nonspecific binding of Hox proteins to beads,untagged WT samples were also subjected to the same regime,and a lack of Hox proteins in bead samples was verified by Western blot(data not shown).Samples for SDS-PAGE were boiled in1ϫLaemmli sample buffer(Bio-Rad),and10l of each was loaded onto precast TGX Stain-free Any kDa gels (Bio-Rad)and transferred onto a PVDF membrane(Bio-Rad). For Western blotting,the SnapID system(Millipore)was used according to the manufacturer’s instructions.Membranes were blocked in5%BSA,1ϫPBST,and primary and secondary anti-bodies were diluted in1%BSA,1ϫPBST.Polyclonal rabbit primary antibodies developed for this study were generated usingϳ20-amino acid C-terminal peptides(␣-HoxE,-F,-U,-Y,and-H(C-terminal);␣-HoxF and␣-HoxY from these prepara-tions are not shown but were used in quantitation of one-di-mensional Western blots)and a HoxH internal peptide(amino acids300–318)that allows recognition of both processed and unprocessed HoxH(YenZym,San Francisco,CA).␣-HoxY and ␣-HoxF antibodies were also generated in rabbit using full-length E.coli expressed protein(SeqLab).␣-Rps1(Agrisera;notshown)and␣-PsaD antibodies(38)were used as loading con-trols for normalization of band intensities for relative protein level quantitation.Secondary incubation was performed with Clean Blot HRP(Pierce),and Cell Biosciences ChemiWest or Pierce Dura Chemiluminescent reagents were used for signal development.Images and quantitation analysis were processed using a Cell Biosciences FluoroCam Q Gel imaging system. Hydrogenase Assays—Cells grown to similar logarithmicgrowth densities(OD730ϭ0.2–0.8)were concentrated toOD730ϭ2.5,and200l was added to600l of assay mixture containing Triton X-100(0.05%,w/v),phosphate buffer(50 m M,pH7),and either methyl viologen(MV;1m M final)or NADH(1m M final)in2-ml HPLC vials sealed with anaerobic septa.The mixture was sparged with argon for10min,100l of anaerobic sodium dithionite(10m M final)was added,and the mixture was incubated with shaking for30min at37°C.The reaction was stopped by the addition of100l of20%(w/v) trichloroacetic acid,and100l of the1-ml headspace was ana-lyzed using a gas chromatograph(Agilent1100)equipped with a Molecular Sieve5A column,a thermal conductivity detector,and argon as the carrier gas.To measure in vivo H2production,cells were spun down and resuspended to OD730ϭ2.5in2ml of BG11containing5m M glucose,placed in a15-ml culture tube, sealed with anaerobic septum,sparged with argon for30min, and incubated at30°C in the dark for24h.A100-l sample from the13-ml headspace was analyzed using a gas chromato-graph as above.Determination of Doubling Times—Log phase precultures (10ml)of WT and hoxϪstrains were used to inoculate400-ml capacity Photobioreactor culture vessels(FMT400,Photon System Instruments)without the addition of antibiotic to growth medium.Illumination parameters were programmed as listed in Table1.Optical density was measured every1–5min at wavelength maximum680nm and recorded digitally,alongwith pH and O2concentration(not shown).Culture vessels were maintained at30°C and continuously bubbled at a rate of ϳ250ml/min in air or argon supplemented with2%CO2.Opti-cal density values versus time were fitted to a single exponential function to determine doubling times.Fluorescence Measurements—Dark-adapted quantum effi-ciency(Fv/Fm)and effective quantum efficiency under illumi-nation(⌬F/FmЈ)of WT and hoxϪstrain suspensions from log phase cultures were measured in multiwell plates with a closed FluorCam(FC800-C/1010,Photon System Instruments)and used to calculate effective electron transport rates.⌬F/FmЈwas determined for cells under continuous illumination for at leastCyanobacterial Hox Hydrogenase Complex Association and Functionat NATL RENEWABLE ENERGY, on January 16, Downloaded from3min at each light intensity with dark adaption between exper-iments.Cells were dark-adapted for at least 5min prior to measurement and were measured in biological and technical triplicate (minimum number of samples per type ϭ9).RESULTSComposition of Hox Hydrogenase Protein Complexes —Re-ported subunit composition and stoichiometry of purified Hox hydrogenase varies in the literature (12,16–20),raising ques-tions about its in vivo structure.Therefore,we assessed frac-tionation of the Hox hydrogenase complex by performing size exclusion FPLC analysis of Synechocystis WT soluble extracts followed by Western blotting with Hox subunit-specific anti-bodies (Fig.1).As expected,all five subunits,HoxE (18.8kDa),HoxF (57.8kDa),HoxU (26.2kDa),HoxY (20.0kDa),and HoxH (52.9kDa),clearly co-fractionate (fractions 10–14).The band intensities of the diaphorase (HoxEFU)and hydrogenase (HoxYH)subunits fluctuate in a related manner,consistent with HoxYH and HoxEFU subcomplexes observed in purifica-tions of other Hox hydrogenases (12,16,17).The presence of what is probably monomeric HoxE (fractions 20–22)suggests that HoxE may dissociate relatively easily from the complex.Interestingly,unprocessed,immature HoxH co-fractionates with other Hox proteins,including processed HoxH (fractions 10and 16–18),implying association of unprocessed HoxH withother complex subunits prior to maturation and full complex assembly (Fig.1,HoxH (open arrows )and HoxH (C-terminal)blots).In addition,unprocessed HoxH is clearly present in higher molecular weight complexes (fractions 8and 9),reveal-ing its association with other unknown proteins (possibly mat-uration factors)prior to its cleavage.Results from size exclusion FPLC imply the presence of Hox complexes and possible subcomplexes,but co-fractionation does not provide enough evidence to confirm subunit associa-tion.To verify Hox protein complex/subcomplex associations in soluble extracts,we performed two-dimensional BN/SDS-PAGE followed by Western blotting (Fig.2).Using this approach,we were able to identify a complex containing all five Hox subunits,again consistent with the composition of the purified pentameric complex reported in the literature (19,20).The apparent molecular mass of this complex is ϳ160kDa,close to the calculated mass based on the predicted sizes of the individual subunits (175kDa).We note that no dimer of this pentameric hydrogenase protein complex could be reliably detected in our analysis despite previous reports (19).However,we were able to detect the presence of HoxFUYH,HoxEFU,and HoxFU subcomplexes.We also failed to reliably detect a HoxYH subcomplex,yet there was a weak antibody signal for the HoxH subunit with a higher apparent molecular massthanFIGURE 1.FPLC and Western blot analysis of WT Synechocystis sp.PCC 6803Hox hydrogenase.The crude soluble fraction of WT Synechocystis sp.PCC 6803was separated by FPLC,and the resulting fractions were analyzed by Western blotting.A ,silver-stained TGX Any kDa gradient SDS-polyacrylamide gel (Bio-Rad)of starting material (Sol .Extr .)and fractions 1–22(for the chromatogram at A 280and A 420see supplemental Fig.1).B–G ,Western blotting of FPLC fractions with the indicated Hox subunit-specific antibodies.Arrowheads in F indicate the signal for unprocessed HoxH.Cyanobacterial Hox Hydrogenase Complex Association and Functionat NATL RENEWABLE ENERGY, on January 16, 2013 Downloaded fromthat of the free HoxH protein alone (Fig.2G ,asterisk ),suggest-ing the association of HoxH with another protein.Monomeric subunits were also detected in the low molecular mass region of the Western blots,with the HoxE,HoxH,and HoxU subunits all accumulating to significant levels when compared with lev-els of each associated with subcomplexes.We were unable to consistently detect unprocessed HoxH in any of these sub-complexes,suggesting that its possible association may only be transient.Nevertheless,these data reveal for the first time the presence of various subcomplexes of Synechocystis Hox hydrogenase in vivo .Subunit Abundance in Hox Hydrogenase Mutants —The detection of Hox subcomplexes in the WT strain by FPLC and two-dimensional BN/SDS-PAGE analyses prompted us to con-struct mutants of each hox gene alone and in combination in the same parental WT strain (supplemental Fig.2)to further examine any relationship between complex/subcomplex asso-ciations and relative subunit abundance by Western blotting.All of the mutants except for hox Ϫ(whole operon deletion con-structed by T.Ogawa)and hoxH Ϫ(see “Experimental Proce-dures”and supplemental Fig.2)were created by ORF replace-ment with similar size antibiotic resistance genes to avoid changes in operon transcription from additional promoters in antibiotic resistance cassettes or large alterations in sequencesize.As expected,no detectable Western signal for Hox sub-units was apparent when the respective gene was disrupted,confirming full segregation of gene deletions (cyanobacteria carry multiple genome copies)(Fig.3A ).Interestingly,deletion of hoxE ,the first gene in the operon,led to a 2–3-fold increase in the remaining Hox protein subunits (Fig.3B ).The increase in Hox protein levels in this mutant is probably linked to the first 190bp of the hoxE ORF,whose absence results in increased hox transcript levels and possible enhancement in the rate of trans-lation due to a decrease in minimum free energy in the hoxE Ϫmutant mRNA (see “Discussion”and supplemental Fig.3,A–D ).The overexpression of HoxH in the hoxE Ϫmutant leads to a discernible increase in levels of unprocessed HoxH com-pared with WT and other hox mutants (Fig.3A and supplemen-tal Fig.4),consistent with increased unprocessed large subunit when the hydrogenase is overexpressed without additional maturation factors in previous studies (our unpublished results 3for R.eutropha soluble hydrogenase and Refs.13and 20).All other hox mutants exhibited decreased levels of remain-ing complex subunits (Fig.3B ).Levels of HoxH and HoxY are decreased to 20and 10%of WT in the respective hoxY Ϫand hoxH Ϫmutants,whereas HoxE,HoxF,and HoxU levels are less than 5%of WT in hoxF Ϫand hoxU Ϫmutants,highlighting interdependencies for HoxYH and HoxEFU subcomplex pro-tein abundance.In addition,HoxY and HoxH levels are decreased to 15–90%of WT in HoxEFU subcomplex mutants (most notably in hoxU Ϫmutants),and HoxE,HoxF,and HoxU levels are decreased to 25–70%of WT in HoxYH subcomplex mutants,revealing interdependencies in subunit abundance across subcomplexes as well.Hox Subunit Association in Individual Subunit hox Mutants —Although changes in transcription could be responsible for altered subunit abundance observed in hox mutants,the absence of complex subunits could also lead to instability of the remaining subunits because of an inability to form stable Hox complexes/subcomplexes.To assess the association of the remaining Hox subunits when one of the five subunits is absent,we again performed two-dimensional BN/SDS-PAGE and Western blotting of soluble extracts from single-subunit hox mutants (Fig.4).No notable differences in total protein were observed between the WT and hox mutant strains by Coomass-ie-stained one-dimensional BN or silver-stained two-dimen-sional SDS-polyacrylamide gels (data not shown).In the hoxE Ϫstrain,HoxFUYH and HoxFU subcomplexes accumulated (Fig.4A ),consistent with the observation of those subcomplexes in WT (Fig.2).Despite the increased levels of HoxY and HoxH in our hoxE Ϫstrain (Fig.3A ),we still were not able to observe the expected HoxYH subcomplex (Fig.4A ).Interestingly,none of the Hox subcomplexes are detected in hoxF Ϫor hoxU Ϫstrains,with the remaining Hox proteins observed only as unassembled monomers,indicating that HoxF and HoxU are necessary for stable assembly/maintenance of the full Hox complex and Hox subcomplexes (Fig.4,B and C ).In contrast,the diaphorase sub-units are still able to assemble into HoxEFU and HoxFU sub-complexes in hoxY Ϫor hoxH Ϫstrains (Fig.4,D and E ),con-3C.Eckert,J.Yu,and P.-C.Maness,unpublishedresults.FIGURE 2.Two-dimensional BN/SDS-PAGE and Western blot analysis of WT Synechocystis sp.PCC 6803Hox hydrogenase.A ,the soluble fraction of WT Synechocystis sp.PCC 6803was separated on a 12%(w/v)polyacrylamide BN-polyacrylamide gel followed by two-dimensional separation on a 17.5%(w/v)polyacrylamide SDS-polyacrylamide gel (B )and analysis by Western blotting with the indicated Hox subunit-specific antibodies (C–G ).The first dimension BN-polyacrylamide gel was Coomassie-stained,whereas the sec-ond dimension SDS-polyacrylamide gel was silver-stained.Cyanobacterial Hox Hydrogenase Complex Association and Functionat NATL RENEWABLE ENERGY, on January 16, 2013 Downloaded fromfirming that HoxY and HoxH are not required for the stable association of diaphorase subcomplexes.Pull-down Analysis of Hox Subunit Associations in Hox Hydrogenase Subcomplexes —Additional mutant strains were constructed to ectopically express His 6-tagged Hox subunits to further probe subcomplex associations:1)HoxYH,tagged on HoxY (hox ϪϩHis 6-HoxYH);2)HoxEFUYH,tagged on HoxH(hoxH ϪϩHis 6-HoxH);and 3)HoxEFUYH,tagged on HoxE (hoxE ϪϩHis 6-HoxE).Overexpression of HoxH by ectopic expression (Fig.5,A and B )or in the hoxE Ϫstrain background (Fig.5C )results in clear unprocessed and processed HoxH sig-nal (Fig.5,A–C ,upper and lower bands ,respectively,in HoxH blots and in the HoxH (C-terminal)blot),increasing the likeli-hood that we would see unprocessed HoxH in bead fractions ifFIGURE 3.One-dimensional SDS-PAGE and Western blot analysis of Synechocystis sp.PCC 6803hox mutants.A ,whole cell lysates of WT and individual/combined hox mutants were run on TGX Any kDa gradient SDS-polyacrylamide gels (Bio-Rad),transferred to PVDF,and immunoblotted with Hox subunit-specific antibodies and PsaD and/or Rps1(not shown)as loading controls.B ,relative levels of Hox subunits in each mutant,presented as a percentage of WT.Data represent quantitation of multiple Western blot analyses with error bars depicting variation between separate analyses.Cyanobacterial Hox Hydrogenase Complex Association and Functionat NATL RENEWABLE ENERGY, on January 16, 2013 Downloaded from。
叶绿素荧光成像技术的原理与应用

叶绿素荧光成像技术的原理与应用一、引言叶绿素是植物中最重要的光合色素,是植物进行光合作用的基础。
溶剂化的叶绿素主要吸收蓝色和红色光,在500~600和650~700nm波长范围内,具有两个吸收峰。
叶绿素荧光成像技术是基于叶绿素发出的荧光信号来进行影像测量的一种实时、无创的模拟测量方法。
本文将介绍叶绿素荧光成像技术的原理、实验流程及其应用。
二、原理叶绿素荧光成像技术是基于叶绿素荧光的成像,叶绿素荧光受光强度和环境因素的影响而变化,可以反映植物的生长状态、光合作用效率和叶片生理变化等信息。
叶绿素荧光成像系统具有高时间分辨率、高空间分辨率的特点,可以获取全景、彩色、实时和定量信息。
叶绿素荧光成像技术主要是利用荧光成像仪和其他仪器支持,通过蓝/绿或红/绿激发光、荧光图像采集和分析等步骤,可以获得叶绿素的分布信息。
三、实验叶绿素荧光成像技术的实验主要分为两个步骤:激发和成像。
首先是激发,将叶片放入光合器中,用荧光成像仪对植物叶片进行光激发,根据荧光成像仪的激光幅度,可以调整植物叶片的荧光强度。
之后,进行成像,将植物叶片放到荧光成像仪中进行拍摄,获取叶绿素的发光信号。
最后,通过荧光照片的处理,可以计算叶片荧光强度和叶绿素荧光参数,如最大光化学利用率、植物光合作用效率等。
四、应用叶绿素荧光成像技术的应用非常广泛,主要涉及到生物学、生态学、农业、气象学,特别适用于植物生长状态监测、植物抗性研究、光合作用效率评估等。
一些具体的应用领域可以如下简要介绍:1.光合作用研究叶绿素荧光成像技术可用于研究植物的光合作用效率、光能利用和光保护机制。
典型的光合作用实验是通过比较光照和黑暗条件下植物的荧光变化来确定植物的光合反应和光保护机制。
2.气候变化影响研究在气候变化方面,叶绿素荧光成像技术可用于研究气候变化导致的植物响应和适应。
通过对多个季节的荧光成像分析可以确定气候变化对地上层和植物生长的影响。
3.生态环境研究叶绿素荧光成像技术可用于研究萎缩地区的植被恢复和生态系统的响应。
FlourCam荧光成像系统

仪器使用
• 连接线路 • 连接电脑
1。Usb 2.0 2。部分电脑可能需要专门的usb驱动(可下 载) • 软件安装 可解压直接执行,也可安装使用。
PSI公司软件与相关资料下载地址http://www.psi.cz/ftp/
科研应用
1.光系统II反应中心光化学效率的表征
光化学效率( FV’/Fm’)的变化,反映PSII
反应中心色素
光反应过程
特定波长光能
原初电 接收的光能
子受体
P·A→ P*·A → P+·A-
后 续
原初电子分离
反 应
PSII(700nm)&PSI(680nm)
基态捕光色素
激发态
热能
产生荧光
叶绿素的激发与退激
叶绿素荧光现象
光合作用示意图
蓝光
热
光合
热
红光
荧光
NADP
NADPH
光系统Ⅰ(PSI)能被波长700nm的 光激发,又称P700 光系统Ⅱ(PSⅡ)吸收高峰波长为680nm,又称P680
250 FM
200
150 FV
100
50 F0
0
FM’ FS
-10 0 10 20 30 40 50 60 70 80
TIME, seconds
运用叶绿素荧光技术来表征光 能的利用、传递、耗散,
能够很好地研究植物的光合作 用过程.
Thanks for your interesting and
questions!
光化学淬灭(qp):反映PSII天线色素分子 吸收光能后,用于光化学电子传递的份 额, 因此也反映了色素天线吸收的光能用 于光合电子传递的变化;同时,qp 也反映 PSII初级电子受体(QA)氧化还原状态的变化。 要保持高的光化学淬灭,就要使PSII反应中 心处于开放状态
叶绿素荧光成像技术在植物生长中的应用

叶绿素荧光成像技术在植物生长中的应用叶绿素荧光成像技术,是一种非侵入式的植物生长观测方法。
它可以在不对植物造成任何伤害的情况下,实时地观测植物的光合作用和植物生长状态。
叶绿素荧光成像技术的应用范围十分广泛,包括植物生长研究、环境监测、农业生产等方面。
叶绿素荧光成像技术的基本原理是,利用叶绿素分子在光合作用中产生的荧光信号,来反映叶片的光合效率。
这种荧光信号可以通过特殊的摄像设备,即叶绿素荧光成像仪来采集。
通过对采集到的荧光图像进行处理,可以得到植物的光合作用效率、光能利用率等多项指标,从而揭示植物生长状态和环境条件对植物生长的影响。
在植物生长方面,叶绿素荧光成像技术的应用主要集中在三个方面:一、对不同生长环境下的植物进行光合作用效率观测。
利用叶绿素荧光成像仪可以在植物生长中实时地观测其光合作用的运作情况。
通过在不同环境和条件下对植物进行观测,可以更加准确地了解植物生长的条件和需求,为生产和研究提供参考。
二、对不同植物的生长状态进行监测。
叶绿素荧光成像技术还可以用于对不同植物的生长状态进行监测,从而判断不同的生长阶段、生长速度等。
这对于农业生产和植物育种方面都具有很大的意义,可以指导地面管理、育种选材等方面的工作。
三、对不同生物模型进行生长动态分析。
除了对植物进行观测之外,叶绿素荧光成像技术还可以用于对其他生物模型的生长状态进行监测。
例如,可以将该技术应用于对微生物、食品发酵过程等生物模型进行生长动态分析,从而更好地了解生物系统的生成规律和规律变化,为相关研究提供参考。
总之,叶绿素荧光成像技术的应用具有非常广泛、多样化的特点。
通过该技术可以实时地观测不同生境下植物的生长状态,从而更好地了解植物的光合作用效率、生长阶段等内容。
这对于农业生产、生物育种和环境监测都具有很大的实用价值。
因此,该技术的发展和应用前景十分广阔。
植物叶绿素荧光成像技术在国内的应用

植物叶绿素荧光成像技术在国内的应用(第四期)植物叶绿素荧光成像技术作为最早实用化的叶绿素荧光成像技术,是目前世界上最权威、使用范围最广、种类最全面、发表论文最多的叶绿素荧光成像技术。
涵盖了从叶绿体、单个细胞、微藻到叶片、果实、花朵,乃至整株植物和植物灌层,几乎可以测量所有的植物样品,甚至包括含有叶绿素的微生物和动物。
叶绿素荧光成像技术最早在21世纪初引进到国内,但一直到2010年后国内的科学家才在国际交流中逐渐发现这项技术的巨大价值,在短短数年中也利用这一技术发表了几十篇高水平SCI 文献。
本期主要介绍目前叶绿素荧光成像技术在国内的应用情况。
一、 植物光合生理研究叶绿素荧光可以直接反应植物光系统的生理状况,因此从叶绿素荧光技术发明之初,就被用于各种植物光合生理研究。
山东农科院使用FluorCam 叶绿素荧光成像技术研究了小麦旗叶与外露花梗光合能力的差异[1]。
研究中发现在小麦生长前中期,旗叶与外露花梗的最大光化学效率Fv/Fm 和量子产额ΦPSII 基本相同。
但在生长后期,旗叶的光合能力显著下降,而花梗光合能力的下降幅度要小于旗叶(图1)。
这证明了在生长后期的灌浆期,花梗对维持籽粒的生长更为重要。
之后,他们又研究了小麦叶片和颖片季节衰老过程中以及颖果发育过程中光合特性的变化[2;3,图2]。
图2. 不同生长期小麦叶片和颖片的最大光化学效率Fv/Fm (A )、量子产额ΦPSII (B )和非光化学淬灭NPQ (C )的变化图1. 不同生长阶段的旗叶(A ,C )和外露花梗(B ,D )的Fv/Fm (A ,B )和ΦPSII (C ,D )典型叶绿素荧光成像图二、植物生物/非生物逆境胁迫与抗逆性研究由于几乎所有种类的生物/非生物逆境胁迫都会影响到植物光合系统的正常生理功能,而叶绿素荧光技术是公认的植物逆境光合功能研究最灵敏的无损探针。
因此通过叶绿素荧光成像技术不但能反映植物受胁迫程度和抗逆能力的差异,而且能指明胁迫影响光合系统的具体机理过程。
FluorCam叶绿素荧光成像一

FluorCam
荧 光 成 像 : 荧 光 淬 灭 分 析
JIP-‐test(电子传递抑制剂敌草隆对OJIP的影响
叶绿素荧光技术著名厂商
• PSI:捷克Brno(孟德尔在此实验并发现著名的孟德 尔遗传定律),Ladislav Nedbal为首席科学家和主要 股东(另一股东为David Kramer,美国密执根州立 大学教授),1997年为美国华盛顿大学H. Pakrasi教 授研制成了第一台FluorCam荧光成像系统。主要产 品有
Kautsky effect
(资料源自Ecolab实验室荧光成像数据库)
荧光淬灭分析(Quenching Analysis)
1. Fv/Fm, 最大光量子产量 2. Fq’/Fm’,Genty参数、ϕPSII,又称光量子效率,用于光化学过程分析评估, 表示PSII吸收的光辐射用于光化学过程的比率,不需要暗适应,但受自然 光照影响大 3. Fv’/Fm’,开放PSII反应中心的光量子效率,不需暗适应 4. Fq’/Fv’,光化学淬灭qP,大致相当于初级受体QA的再氧化状态 5. (Fm-‐Fm’)/Fm’,非光化荧光淬灭
显微叶绿素荧光成像系统
• 可对植物组织、藻类等细胞或亚亚细 胞结构进行荧光成像测量 • 便携式标准版显微荧光成像系统, Olympus CX31显微镜 • 便携式增强版显微荧光成像系统, Olympus BX40 显微镜及可扩展配件 (通过扩展可测量GFP等) • 多功能显微荧光成像系统,除具备 Olympus BX40 显微镜及可扩展配件外, 还具备6位滤波轮(有机械调节和电 子调节两种模式供选配),因而除可 以成像测量叶绿素荧光外,还可测量 GFP、CY3、CY5等多种稳态荧光或生 物荧光
FluorCam荧光成像系统Protocols(实验程序)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(2012年5月中科院植物所 叶绿素荧光技术及其应用研习班) 易科泰生态技术公司 Ecolab生态研究室
FluorCam叶绿素荧光成像技术及其应用
• • • • • 叶绿素荧光技术 FluorCam叶绿素荧光成像 FluorCam叶绿素荧光成像应用案例 Ecolab实验室叶绿素荧光成像实验 FluorPen手持式叶绿素荧光仪与AquaPen 藻类荧光仪
FluorCam荧光成像技术特点
• • • • • • • • • • • 高灵敏度CCD镜头,时间分辨率可达每秒50张,快速响应和实验程序分析,精 确反映叶绿素荧光瞬变 可选配高分辨率(高像素值)CCD镜头,适于需要高分辨率荧光成像分析如 GFP等稳态荧光 软件具备Fv/Fm、Kautsky 诱导效应、荧光淬灭分析、稳态荧光成像、PAR 吸 收指数、LC等自动实验程序,实验程序可设置更改,或创建新的实验程序 具备实验程序自动重复功能,从而实现无人职守自动成像实验 “4+1”LED光源(周边4个LED光源板,顶部一个LED光源板),可选配红色光 源、橙色光源、绿色光源、青色光源、蓝色光源、白色光源、红外光源、紫 外光源等各种波长光源,光强可达10000μmol/m2.s 可选配单周转光闪(STF),100μs光闪(光脉冲)强度达120000μmol/m2.s 可选配8位滤波轮 可选配QA再氧化动力学测量模块 可选配OJIP快速荧光动力学测量模块 可选配可见光真彩成像分析模块 可选配植物呼吸与光合作用测量模块
FluorCam开放式叶绿素荧光成像系统
标准版成像面积可达13x13cm,大型版可达20x20cm, 镜头光源可灵活调整
FluorCam移动式植物荧光成像系统
• 整套系统装在有4个轮子的 支架上,方便野外移动 • 镜头及光源高度可以在 20-‐150cm范围内调节 • 可选配暗适应箱 • 成像面积20x20cm • 可附加镜头用于真彩成像分 析
样带扫瞄式植物荧光成像系统与 三维立体植物荧光扫瞄成像系统
• 样带荧光成像系统可以对20-‐200cm或 100x200cm范围的样方植物荧光扫描成像,以 便于梯度胁迫研究等 • 立体荧光成像系统可以通过对整株植物叶绿 素荧光扫描成像,并通过软件形成3维立体荧 光成像
多光谱XY-‐平台式大型植物荧光成像系统
显微叶绿素荧光成像系统
• 可对植物组织、藻类等细胞或亚亚细 胞结构进行荧光成像测量 • 便携式标准版显微荧光成像系统, Olympus CX31显微镜 • 便携式增强版显微荧光成像系统, Olympus BX40 显微镜及可扩展配件 (通过扩展可测量GFP等) • 多功能显微荧光成像系统,除具备 Olympus BX40 显微镜及可扩展配件外, 还具备6位滤波轮(有机械调节和电 子调节两种模式供选配),因而除可 以成像测量叶绿素荧光外,还可测量 GFP、CY3、CY5等多种稳态荧光或生 物荧光
FluorCam便携式叶绿素荧光成像仪
最大成像面积3.5x3.5cm, 备选配件包括叶夹、三脚架、便携蓄电池等
FluorCam封闭式叶绿素荧光成像系统
成像面积可达13x13cm, 可选配QA再氧化测量模块、OJIP测量模块等
GFPCam封闭式多光谱荧光成像系统
• 配有8位滤波轮,除具有封闭 式叶绿素荧光成像系统的功能 外,还可以成像测量绿色荧光 蛋白(GFP)等其它稳态生物 荧光 • 成像面积可达13x13cm • 可选配QA再氧化动力学测量模 块,OJIP快速荧光动力学测量 模块
– FluorCam叶绿素荧光成像系列产品 – FL3500/FL5000双调制荧光仪系列产品 – FluorPen及AquaPen等手持式荧光仪产品 – 光养生物反应器等藻类培养与在线监测产品 – 光源与植物培养室
• Walz:德国,主要产品为PAM2500叶绿素荧光仪等 • OpKcs:美国,主要产品为OS5p-‐PAM叶绿素荧光仪 等
– Ulrich Schreiber, Pulse-‐Amplitude-‐ModulaKon(PAM) Fluorometry and SaturaKon Pulse Method, An Overview – Ladislav Nedbal, Chlorophyll Fluorescence Imaging of Leaves and Fruits
FluorCam叶绿素荧光成像
1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Handy FC——便携式叶绿素荧光成像仪 Handy GFPCam——便携式荧光成像仪 Handy Leaf-‐chamber——便携式光合联用叶绿素荧光成像系统 Open FC——开放式叶绿素荧光成像系统,有13x13cm和20x20cm两种配 置方案 Closed FC——封闭式叶绿素荧光成像系统 Closed GFPCam——封闭式多光谱荧光成像系统 Micro-‐FluorCam——显微叶绿素荧光成像系统,又分标准版、增强版 (可选配GFP Filter Cube Set)及滤波轮版 Rover FluorCam——移动式大型植物荧光成像系统 Transect FluorCam——样带扫描式植物荧光成像系统 XY-‐Plane FluorCam——多光谱XY-‐平台式大型植物荧光成像系统 Conveyor and RoboKc PlantScan System——PlantScan全自动植物光谱成像 分析系统 Arch FluorCam——拱形三维植物荧光扫描成像系统 Fluorescence KineKc Microscope——FKM荧光动态显微光谱成像系统
叶绿素荧光技术发展
• Kautsky effect: Kautsky and Hirsch (1931) 首次用肉眼发现 叶绿素荧光现象并发表论文“CO2同化新实验”,后被 称作“Kautsky effect” • PAM (Pulse Amplitude Modulated Fluorometer): Schreiber(1986)等发明了PAM脉冲调制技术测量叶绿 素荧光 • FluorCam:KineKc imaging of chlorophyll fluorescence: Ladislav Nedbal(2000)等于上世纪90年代末期发明了 与PAM技术相结合的叶绿素荧光成像技术 • Chlorophyll a Fluorescence, A Signature of Phptosynthesis(Papageorgiou and Govindjee, 2009)
Kautsky effect
(资料源自Ecolab实验室荧光成像数据库)
荧光淬灭分析(Quenching Analysis)
1. Fv/Fm, 最大光量子产量 2. Fq’/Fm’,Genty参数、ϕPSII,又称光量子效率,用于光化学过程分析评估, 表示PSII吸收的光辐射用于光化学过程的比率,不需要暗适应,但受自然 光照影响大 3. Fv’/Fm’,开放PSII反应中心的光量子效率,不需暗适应 4. Fq’/Fv’,光化学淬灭qP,大致相当于初级受体QA的再氧化状态 5. (Fm-‐Fm’)/Fm’,非光化荧光淬灭
பைடு நூலகம்
FluorCam
荧 光 成 像 : 荧 光 淬 灭 分 析
JIP-‐test(电子传递抑制剂敌草隆对OJIP的影响
叶绿素荧光技术著名厂商
• PSI:捷克Brno(孟德尔在此实验并发现著名的孟德 尔遗传定律),Ladislav Nedbal为首席科学家和主要 股东(另一股东为David Kramer,美国密执根州立 大学教授),1997年为美国华盛顿大学H. Pakrasi教 授研制成了第一台FluorCam荧光成像系统。主要产 品有
• 测量系统在柜体内, 光源及摄像头高度和 位置可调 • 测量面积40x80cm或 40x100cm供选择 • 可用于培养箱样品多 光谱分析
PlantScan全自动植物光谱成像分析系统
• 高通量植物成像观测系统,具备植 物传送系统、植物称重系统、自动 浇灌系统等 • 可配备温湿度及光照自动控制系统 • 植物荧光成像与可见光真彩成像, 植物荧光成像面积30x35cm • 可选配植物热成像模块 • 可选配植物红外成像模块 • 可选配植物VNIR高光谱成像分析模 块,光谱范围400-‐1000nm,成像面 积30x35cm;或选配SWIR高光谱 (1000-‐2500nm)成像
FKM荧光动态显微光谱成像系统
• 由显微荧光成像、双调制荧光测量及光谱仪组成 • 可同步荧光成像和点快速荧光动力学测量 • 可通过选择不同的激活光谱激活不同部位的天线色素;可用于分析 哪些色素蛋白复合体对光化学荧光淬灭或非光化学荧光淬灭贡献最 多 • 可用于活体检测非叶绿素荧光动态,如生物自发光或荧光颜料等, 并与同一细胞的叶绿素荧光动态相比较 • 可用于检测分析QA再氧化、连通性、天线大小等快速过程
叶绿素荧光技术常用参数
• Fo、Fm与QY,此外还有PAR_Abs及ETR • Kautsky诱导效应:Fo,Fp,Fv,Ft_Lss,QY, Rfd • 荧光淬灭分析:Fo,Fm,Fp,Fs,Fv,QY, ΦII,NPQ,Qp,Rfd,qL等50多个参数 • OJIP与JIP-‐test:Fo,Fj,Fi,P或Fm,Mo(OJIP 曲线初始斜率)、OJIP固定面积、Sm(对关闭 所有光反应中心所需能量的量度)、QY、PI等 • 光响应曲线LC:Fo,Fm,QY,QY_Ln
FluorCam荧光成像系统Protocols(实验程序)
Quenching(荧光淬灭分析) Kautsky inducKon(Kautsky诱导效应) Light curve(光响应曲线) PAR absorpKvity 实验程序自动重复功能 QA reoxidaKon OJIP,Fast fluorescence inducKon with 1μs resoluKon Measured parameters: Fo, FM, Fv, Fo’, FM’, Fv’, QY(II),more than 50 calculated parameters • GFP等静态荧光 • NDVI • • • • • • • •