化学结构与药理活性
药物的化学结构和药效的关系

离子通道( ion channel )
绝大多数通道蛋白形成的与离子转运有关 的有选择性开关的多次跨膜通道.
特点
一 具有离子选择性,离子通道对被转运离 子的大小与电荷都有高度选择性,而且转运 速率高,其速率是已知任何一种载体蛋白的 最快速率的1000倍以上.
2021/1/12
16
二 离子通道是门控 离子通道的活性由通
NO2 ≥ COOH > COCH3> CHO > OH >
NHCOCH3> NH2 > CONH2 > SO2NH2
2021/1/12
40
2、解离度对药效的影响
□ 有机药物多数为弱酸或弱碱,在体液中只 能部分离解
□ 药物的离子型和分子型在体液中同时存在
□ 通常药物以分子型通过生物膜,进入细胞 后,在膜内的水介质中解离成离子型,以 离子型起作用。
□ 与药物结构、理化性质密切相关
□ 其作用与体内特定的受体相互作用有关
□ 同一药理作用类型的药物与某一特定的受体 相结合,在结构上往往具有某种相似性
□ 同类药物中化学结构相同的部分称为该类药
2021/1物/12 的基本结构(药效结构)
3
药物和受体的相互作用
2021/1/12
4
药物效应动力学
1 药物的基本作用
1 药物的基本作用
22
药物的作用靶点
3 药物的作用机制
44
药物与受体
■以受体为靶点 ■以酶为靶点 ■以离子通道为靶点 ■以核酸为靶点
2021/1/12
7
药物效应动力学-受体
2021/1/12
8
常见与受体有关的药物
受体
药物
化学结构与药理活性
非解离%
解离%
2.0
100.0
0.00
4.0
99.96
0.04
6.0
96.17
3.83
7.0
71.53
28.47
8.0
20.02
79.93
10.0
0.25
99.75
12.0
0.00
100.0
第二章 化学结构与药理活性 第一节败涂地 化学结构与理化性质(Structure Activity Relationship Pharmacokinetic Phase)
3、胎盘屏障和药物分布
第二章 化学结构与药理活性 二、影响药物到达作用部位的因素 (三) 药物的蛋白结合
药物进入血液后与血浆蛋白的结合是影响药物分布、代谢和排泄的重要因素。 药物与血浆蛋白的结合特点:
1、可逆的结合(多数是以氢键、范德华力、疏水键、离子键结合的); 2、结合物不能通透生物膜(药物-蛋白结合物分子量大); 3、药物-蛋白结合物没有药理活性;
药物大多数为有机弱酸和弱碱(见P10,表2-3),在体液中存在着解离平衡。 只有未解离型的药物才能通透脂质的生物膜。 即有机弱酸、弱碱药物的吸收与它们的解离度有关。 那么解离度又与什么有关呢?
二、药物的解离度
解离度与什么有关?
药物的解离度与它的解离常数pKa有关,与药物所处的体内介质的pH有关。 对于酸性药物有: pKa:药物的解离常数; pH:介质的pH。 酸性药物在pH小的介质中,解离度小,未解离型药物浓度高。
04
03
01
02
第一节 化学结构与理化性质
药物的脂水分配系数对药物吸收的影响
疏水常数л具有加和性,即化合物分子的分配系数
药物的化学结构与生物活性的关系

系
第一节 定义和范围
医学ppt
1
(1)反映药物作用的特异 性
构效关系
structure-activity
relationship(SAR)
化学结构与生物活性 (药理,毒理)之间的关 系
(2)有助于解析,认识药物的
作用机理 (mechanism of action)和作用方式(mode of action)
医学ppt
苯甲酸酯对局麻 具有重要作用
13
NH2 O
O
苯佐卡因
NH2
O OH
O
奥索卡因
OH O
NH2 O
新奥索仿
溶解度小,不能注射
引入碱性胺側链(类似爱康宁中N)
NH2
O N
Procaine
O
1904年上市,确定了苯甲酸酯类局麻药的诞生
医学ppt
14
其他例子:
R1,R2,R3
O R2 R1
Oห้องสมุดไป่ตู้
R3 N
O N
H
巴比妥类催眠药
良好的脂溶性 分子形式 pKa
决定进入脑内药物量
巴比妥酸 苯巴比妥酸 苯巴比妥 戊巴比妥
pKa
4.12
未解离百分率 0.05
3.75
7.40
0.02
50
8.0 79.92
医学ppt
15
O C2H5
O
H N
O N
H
苯巴比妥
R1和R2基团碳总数为4~8, 具有很好脂水分配系数
N上引入甲基,酸性下降,脂溶性增加
药物:各种信息易于获得 靶点:信息获得较难 药物-靶点复合物:很难
第二章 化学结构和药效关系

构效关系: 构效关系: 化学结构与生物活性 间的关系, 生物活性间的关系 化学结构与 生物活性 间的关系 , 通常称为构效关系 (Structure-activity relationships, SAR),是药物化学研究 , 的主要内容之一。 的主要内容之一。 药物在体内与特定的受体部位发生相互作用,引发生 药物在体内与特定的受体部位发生相互作用, 物活性, 物活性, 药物的化学结构必须与受体大分子的结构相互匹 药物化学结构改变会引起药理活性发生变化;药物的化 配。药物化学结构改变会引起药理活性发生变化 药物的化 学结构还决定其理化性质,影响药物在体内的吸收、 学结构还决定其理化性质,影响药物在体内的吸收、分布 和代谢。因此药物的生物活性与药物的理化性质有关, 生物活性与药物的理化性质有关 和代谢。 因此药物的生物活性与药物的理化性质有关,即 与药物结构中电子云密度有关(电子因素) 与药物结构中电子云密度有关(电子因素),与药物的立 体化学结构有关(空间因素) 体化学结构有关(空间因素)。
(一)药物吸收
P=C生物相/C水相(Partitiong confficient) P=C有机相/C水相(Partitiong confficient) 是药物对油相和水相溶解度的量度, 是药物对油相和水相溶解度的量度 , 也是药物对水相 和器官组织的相对亲和力的度量。 和器官组织的相对亲和力的度量。 C生物相测量困难,用C正辛醇代替。 代替。 量困难, P=C正辛醇/C水相(Partitiong confficient) 由于药物为有机化合物, 值较大 通常用lgP表示 值较大, 表示。 由于药物为有机化合物 , P值较大 , 通常用 表示 。 药物亲脂性时为正值,亲水性药物为负值。 药物亲脂性时为正值,亲水性药物为负值。
氯雷他定的化学结构与药理活性关系

氯雷他定的化学结构与药理活性关系氯雷他定(Loratadine)是一种第二代抗组胺药物,被广泛用于治疗过敏性鼻炎和荨麻疹等过敏性疾病。
它的化学结构和药理活性之间存在密切的关系,这对于理解氯雷他定的药效和副作用至关重要。
氯雷他定的化学结构是一个氯苯环与一个咪唑环的结合。
这种结构使得氯雷他定具有抗组胺的能力。
组胺是一种重要的生物活性物质,它在机体内广泛分布,参与多种生理过程。
在过敏反应中,组胺的释放是引起症状的主要原因之一。
氯雷他定通过与组胺H1受体结合,阻断了组胺的作用,从而减轻了过敏反应引起的症状。
作为第二代抗组胺药物,氯雷他定相比第一代药物具有更好的选择性和安全性。
第一代抗组胺药物常常会引起嗜睡、口干等副作用,而氯雷他定则相对较少。
这主要归因于氯雷他定的化学结构。
相比于第一代药物,氯雷他定的结构更加特异,选择性地与组胺H1受体结合,减少了对其他受体的影响。
这使得氯雷他定在治疗过敏性疾病时更加安全可靠。
除了抗组胺作用外,氯雷他定还具有一定的抗炎和抗氧化作用。
研究表明,氯雷他定能够抑制炎症介质的释放,减轻炎症反应。
此外,它还能够抑制自由基的生成,减少氧化应激对机体的损伤。
这些药理活性使得氯雷他定在一些炎症性疾病的治疗中具有潜在的应用价值。
然而,氯雷他定的药理活性也带来了一些潜在的副作用。
尽管氯雷他定的选择性较高,但它仍然会对其他受体产生一定的影响。
这可能导致一些不良反应,如头晕、恶心等。
此外,长期使用氯雷他定可能会对肝脏产生一定的负担。
因此,在使用氯雷他定时,应注意遵循医生的建议,避免超量使用或长期使用。
总的来说,氯雷他定的化学结构与药理活性之间存在密切的关系。
其特异的结构使其具有抗组胺、抗炎和抗氧化等多种药理作用。
这使得氯雷他定成为治疗过敏性疾病的有效药物。
然而,我们也应该注意其潜在的副作用,合理使用氯雷他定,以确保药物的安全性和有效性。
第三章化学结构与药理活性

第三章化学结构与药理活性化学结构与药理活性是药物研发过程中的一个重要环节。
化学结构主要指的是药物分子的化学组成和结构排列方式,而药理活性则是药物分子与生物体内靶标的相互作用所产生的生理或药效活性。
在药物研发中,了解药物的化学结构对于预测和理解其药理活性至关重要。
药物分子的化学结构决定了其在生物体内的吸收、分布、代谢和排泄等性质,进而影响其药效和毒性。
化学结构的一些重要特征,如官能团的位置和性质、分子的立体结构、化学键的性质等,都可以通过合理设计来调控药物的性质。
例如,对于一些药物来说,引入特定的官能团可以增强其生物活性,而化学键的构型和电子云分布则可以影响药物与靶标之间的相互作用。
药理活性是药物的生物学效应,也是药物的核心功能。
药理活性可以通过与生物体内的靶标结合,调控细胞信号传导通路,从而产生治疗效果。
药理活性的机制可以是激活或抑制特定的受体、调节酶活性、影响细胞内信号传递等等。
药理活性主要通过与靶标的亲和力和选择性来实现,药物与靶标之间的结合可以发生物理、化学或生物学反应,从而改变靶标的功能状态。
药物研发过程中,通过分析药物的化学结构可以揭示其与靶标之间的相互作用模式,预测其药理活性。
例如,根据一些药物结构中特定官能团的存在,可以推测该药物可能具有抑制其中一特定酶的活性。
此外,通过对已知药物的化学结构与活性的大数据分析,可以建立结构-活性关系模型,从而快速预测新药物的活性,加速药物研发过程。
化学结构与药理活性之间的关系还可以用于药物优化。
通过结构修饰和合理的药物设计,可以调节药物分子的化学特性,改善药物的吸收、分布、代谢和排泄性质,提高药物的靶向性和药效。
优化药物的化学结构可以对其药理活性进行改进,增强药物的疗效,并降低不良反应的发生率。
药物化学化学结构和药理活性

资料仅供参考,不当之处,请联系改正。
二、影响药物到达作用部位的因素
• 主要受两大因素的制约. • 一是药物分子因素,即药物的化学结构及由化学结
构所决定的理化性质,如溶解度、分配系数、电离 度、分子间力、氧化还原电位、电子等排、官能 团之间的距离和立体化学. • 二是药物在其中运行的生物学因素,包括药物分子 与细胞间及细胞内体液和与生物聚合物等的相互 作用,这种相互作用决定了药物的吸收,分布和消除 特征,决定了药物的生物利用度.
资料仅供参考,不当之处,请联系改正。
资料仅供参考,不当之处,请联系改正。
三 药物的水溶性
1 氢键 羟基和亚胺基团;可生成的氢键越多,分子的水溶性
越大。
2 解离 离子-偶极键
3水溶性的预测 (1)经验法 有机功能基的碳增溶势
(2)分配系数分析法
分配系数也能预测药物的水溶性。药典关于水溶度的定义,溶解度 大于3.3%为溶解,相当于lgP指0.5。因此以0.5为基准,小于0.5的为 水溶性,大于0.5的为水不溶性的。
第一节
资料仅供参考,不当之处,请联系改正。
化学结构与理化性质
• 一、药物的分配系数
C org
P=
Cw
Corg表示药物在生物非水相或正辛醇中的浓度 Cw表示药物在水相的浓度
• 是评价药物亲脂性或亲水性大小的标准, 即药物在生物非水相中物质的量浓度Corg与 在水相中物质浓度Cw之比。
• 常用其对数lgP表示
• 药物蛋白结合分为可逆和不可逆。在不可逆反 应中,药物通过共价键和蛋白结合。大多数药 物与蛋白的结合时可逆过程,药物以氢键,范 德华力,疏水键和离子键与蛋白结合。
资料仅供参考,不当之处,请联系改正。
化学结构与药理活性

抗肿瘤药氮芥(双氯乙胺烷化剂),在体内能转变成高度活泼的亲电
性的乙烯亚胺,与癌细胞和正常细胞中许多细胞组分,如羟基、巯基、 羧基、磷酸酯和咪唑基发生亲核反应,尤其是将DNA中鸟嘌呤7位氮
烷基化,致使密码错编(Miscoding),最终导致细胞死亡。
Structurely specific drugs
药效团可分两种类型:一类是具有相同药理作用的类似物,它们具
有某种基本结构。在各论中,几乎都有这种类似物的例子;
另一类是一组化学结构完全不同的分子,但它们可以与同一受体以 相同的机理键合,产生同样的药理作用。
有抗药性的原因。
二 、化学结构与药理活性
Chemical Structure-Activity Relationship
(一)药效团(pharmacophore)
在药物-受体相互作用生成复合物过程中,第一步就是药物与受体 的识别。受体必须去识别趋近的分子是否具有结合所需的性质。这 种特征化的三维结构要素的组合称为药效团。
19世纪末至20世纪初,著名微生物家Ehrlich发现,一
些有机物能以高度的选择性产生抗微生物作用,他认为这
是由于药物与生物中某种接受物质结合的结果,提出了接 受物质 (Receptive substance) 和受体 (Receptor) 这些词汇, 并认为药物与受体的相互作用与钥匙和锁相似,具有高度 的契合专一性。
应,从而抑制了酶的再生 。
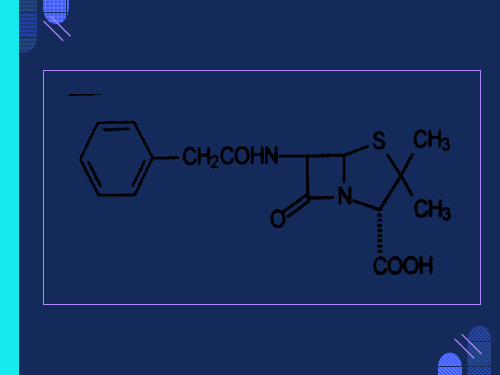
综上所述,PBP是β-内酰胺抗生素的受体,从这个意义上说,细菌 的细胞壁仅仅是抗生素作用的一个特殊部分,实质性的作用还是负 责交联肽聚糖的转肽酶的抑制。
许多细菌能产生β-内酰胺酶,将β-内酰胺环开裂,使抗生素失活。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
给药途径
效应
第一节 化学结构与理化性质
❖ 一、药物的分配系数(Partition coefficient) ❖ 二、药物的解离度(Degree of ionization) ❖ 三、药物的水溶性(Soubility of drugs)
一、药物的分配系数(Partition coefficient,P)
= pKa -pH
❖ 对某一酸性药物而言,环境pH值越小(酸性越 强),则未解离药物[HA]浓度越高。
碱性药物
B + H2O
BH+ + OH-
lg [B] [BH+]
=
pH -
pKa
❖ 对于某一 碱性药物:环境的pH值越大即碱性越强, 则未解离药物[B]浓度越高。
三、药物的水溶性
❖ (一)氢键 ❖ 能够给予或接收氢键的功能基会增加分子的亲水性,而不 能生成氢键的功能基会增加分子的疏水性。 ❖ 通常是,药物可生成的氢键越多,分子的水溶性越大。 ❖ (二)解离 ❖ 有机盐解离成离子,与水分子形成离子-偶极键,从而可 能增加水溶性。 ❖ 一般强酸-强碱、强酸-弱碱、弱酸-强碱生成的盐能完 全解离,与水分子相互作用。弱酸-弱碱生成的盐不能完全解 离,因而没有很好的水溶性。分子内形成离子键,就不能与水 分子作用,则不溶于水。
第二章 化学结构 与药理活性
Chemical Structure and Pharmacologic Activity
化学结构与药理活性
❖ 药物从给药到产生药效是一个非常复杂的过程。 ❖ 随着对药物作用机理研究的深入,人们建立化学结
构与药理活性之间的直接关系是十分艰巨的。 ❖ 将药物在体内作用过程分为三个阶段,容易建立化
Ar
磺胺
RSO2NH2 9~10 磺酰亚胺 RSO2NHCO 5~6
R’
二酰亚胺 RCONHCOR’ 9~10 脂肪酸 RCOOH 5~6
硫醇
RSH 10~11 芳酸 ArCOOH 4~5
苯硫酚
ArSH 9~10
磺酸
RSO3H 0~1
碱性化合物的结构类型及其pKa
碱
结构式 pKa
碱
结构式
pKa
芳基胺 ArNH2 4~5 脂肪胺 RNH2 10~11
❖ 了解药物在体内的转运过程,可以认识药物的构效 关系,进而从各种途径优化药物的生物利用度 (Bioavailability) 。
组织 (Tissue)
分布 Distribution
肌肉或皮下注射
药 静脉注射 物
消化道 肝
血液(Blood) 药物(Drug) [游离型] [结合型]
作用部位 (受体)
❖ 口服吸收时应考虑首过效应对药效的影响(如硝酸 甘油)。
肝肠循环
(enterohepatic cycle)
❖ 一些药物自肝脏分泌到胆囊并排 放到小肠中,小肠又将药物吸收 经门静脉再到达肝脏,这个过程 叫肝肠循环。该循环直到药物在 肝脏中代谢或经肾排泄完为止。
❖ 药物或其代谢产物自胆汁分泌到 小肠,大多与甘氨酸、硫酸氢酯 或葡萄糖醛酸相结合,生成游离 的药物,以致又被吸收入血液中。 例如氯霉素葡醛酸轭合物在胆汁 中被分泌到肠中,经肠中细菌酶 水解后,生成的氯霉素又被吸收。
(一)药物吸收 (Drug absorption)
❖ 1. 药物的分配系数(Partition coefficient) ❖ 2. 药物的解离度(Degree of ionization) ❖ 3. 药物的溶解度 ❖ 4. 药物的分子量
1. 药物的吸收与lgP关系 The relationship of absorption and lgP ofdrugs
π的作用
❖ π具有加和性。
lgP= lgPH + ΣXi
❖ 当脂肪链有分支、成环及双键等因素,还须加上校 正值,分别为-0.20、-0.09及-0.30。
布洛芬
CH3 CH3
CHCH2
CH3 CH COOH
❖ lgP计算=1苯+6甲基+1羧基+2分支 =2.13+3.0+(-1.26)+(-0.40) =3.47
用的强度降低,作用持续时间延长。
❖ 而超短时镇静催眠药硫喷妥在生理pH7.4时的lgP=
2,静脉注射几分钟即穿越血脑屏障,从而迅速催
眠,但也容易经过再分布,积累于脂肪和肌肉中, 使作用持续时间短。
二、影响药物到达作用部位的因素 Factors which drugs arrive action position
❖ 药物分子因素
由药物的化学结构与由结构决定的理化性质,包 括溶解度、分配系数、 解离度、分子间力、氧化 还原电位、电子等排、官能团之间的距离和立体 化学。 ❖ 药物在转运过程中的生物学因素 包括药物分子与细胞间及细胞内体液、与生物聚 合物等相互作用。
❖ 药物在生理pH7.4时,脂溶性(lgP)越大,解离程 度越小,越容易通过。
一些药物的分布情况
药物在血浆与脂肪中分布
❖ 药物在血浆与脂肪之间的分布,取决于lgP。 ❖ lgP 越大,在脂肪中分布越多。因此这种分布影响
药物的作用强度(Potency)和持续时间(Duration)。 ❖ 一般药物的lgP 越大,药物在血浆中浓度越小,作
❖ 药物的分配系数指药物在生物相中物质的量浓度与在水相 中物质的量浓度之比。
C生物相 P=
C水相
Co P o/w =
Cw
❖P值越大,则药物的脂溶性越高。
❖由于药物P值差别较大,所以药物分配系数常用其对数lg P 表示。
疏水常数(π,Hydrophobic constant)
❖ 药物的分配系数取决于它们的化学结构。 由于药物的可看成各取代基按一定方式组合,可 以用疏水常数(Hydrophobic constant) π来表达取代 基的疏水性。
❖ (一)酸碱性 ❖ 药物的酸碱性是根据Bronsted-lowry理论判断,能
产生质子的物质即为酸,能接收质子的物质为碱。 ❖ 药物的酸碱性直接影响药物的药动学行为,非常重
要。 ❖ (二)相对酸性强度pKa
酸性化合物的结构类型及其pKa
酸
结构式
pKa
酸
结构式 pKa
酚
ArOH
9~11 N-芳基磺胺 RSO2NH 6~7
❖ 肝肠循环是药物长效原因之一, 也因此引起药物蓄积中毒。
生物利用度(bioavailability)
❖ 由于药物未必能够完全吸收,用进入血液循环中药 量的份额和吸收的速率,表征药物被机体吸收的程 度,这就是生物利用度。
❖ 药物的化学结构和物理化学性质是决定生物利用度 的主要因素,但难溶物质的颗粒大小,制剂形式和 质量也会影响生物利用度。
第二节 药动相的构效关系 Structure-activity relationship in the parmacokinetical
phase
一、药物的转运
❖ 药物动力相涉及药物从用药部位,经随机运行,到 达最终作用部位的全过程。
❖ 药物经历这样一个转运过程,最后只有一部分药物 到达作用部位。
学结构与药理活性之间关系。
药物作用过程的三个阶段
过程 分类
药剂相
Parmacentical phase
药动相
Parmaco-
kinetic phase
药效相
Parmaco-
dynemic phase
发生 过程
药物的释放 吸收、分布和消 药物-受体在靶组 除(代谢及排除) 织的相互作用
研究 目的
优化处方和 优化生从血液循环进入组织 或器官后,才能发挥药理作用。例如作用于中枢 神经系统药物应分布到中枢;抗肿瘤药物转运到 肿瘤组织中。
❖ 药物与机体的各种组织的亲和力是不同的。因此 药物的组织分布必然对其生物活性产生巨大的影 响。
❖ 药物在体内各组织的分布在很大程度上取决于药 物的理化性质。
❖ 药物的lgP值越大,则药物的脂溶性越高。 ❖ 一般来说,脂溶性药物易吸收。非解离药物吸收与
亲脂性密切相关;可解离药物则与其中未解离分子 的亲脂性有关。
❖ 由于生物膜具有双脂质层的特殊结构,要求药物吸 收的主要条件是适宜的脂溶性和水溶性。
体内不同部位所需的lgP不同
❖ 胃肠道吸收:lg P = 0.5~2.0
研究影响药物到达作用部位的因素
❖ (一)药物吸收( Absorption of drug) ❖ (二)药物向生物作用部位的分布(Distribution) ❖ (三)药物的蛋白结合(Protein binding of drug) ❖ (四)药物从体内消除(Elimination of drug)
吸收
代谢
排泄
(Absorption) (Metabolish) (Excretion)
胃肠道、皮下
尿、胆汁、
肌肉等部位 首过效应 肺等部位
蛋白结合 Protein
重吸收 Binding
肾小管、 肝肠循环
药理 作用
首过效应(first pass effect)
❖ 药物自小肠吸收进入血液循环,首先经门静脉进入 肝脏,肝脏是机体对内源性和外源性物质代谢的主 要器官,因而有相当一部分(甚至全部)药物分子 被代谢,使药物活性降低,这种过程称为首过效应。
氮杂环
5~6 脒
N
R
亚胺 RCH=NH 3~4
胍
NH 10~11
R NH2
NH R N NH2
H
10~11
生理条件下中性的化合物类型
脂肪醇 酯
醛和酮 醚 腈
二芳基胺 四级铵
氧化胺 酰胺 硫醚 亚砜
砜 硫酸酯
环丙沙星
中性 O
F
COOH 酸性
N