埃索美拉唑镁欧洲药典质量标准色谱图
埃索美拉唑镁肠溶胶囊含量测定研究

埃索美拉唑镁肠溶胶囊含量测定研究许剑峰;陈涛【摘要】目的:建立胃食管反流性疾病(GERD)药物埃索美拉唑镁肠溶胶囊活性成分埃索美拉唑镁的含量测定方法.方法:拟采用HPLC法.色谱柱C8键合反相柱,Phenomenex(150×4.6mm,5μm);流动相系统为等度洗脱:乙腈:磷酸盐缓冲溶液(7∶13);检测器为UV检测器,波长280nm;流速为1.0ml/min,进样量为20μl,柱温为30℃.结果:该药活性成分埃索美拉唑镁(以奥美拉唑RS计)进样量在0.80576μg~4.83456μg范围内与峰面积呈良好的线性关系A=15.191C+22.733 (r=0.9997),高中低三个浓度9个样平均加样回收率为100.11%,RSD为0.59%;结论:HPLC法测定埃索美拉唑镁含量准确度高、重复性好,可以作为埃索美拉唑镁肠溶胶囊含量测定方法.【期刊名称】《黑龙江科技信息》【年(卷),期】2016(000)017【总页数】1页(P34)【关键词】埃索美拉唑镁;HPLC法;含量测定【作者】许剑峰;陈涛【作者单位】哈药集团三精制药有限公司,黑龙江哈尔滨150000;哈药集团三精制药有限公司,黑龙江哈尔滨150000【正文语种】中文埃索美拉唑镁为奥美拉唑的左旋异构体,临床上用于胃食管反流性疾病(GERD)-糜烂性反流性食管炎的治疗。
已经治愈的食管炎患者防止复发的长期维持治疗。
胃食管反流性疾病(GERD)的症状控制。
与适当的抗菌疗法联合用药根除幽门螺杆菌,并且-愈合与幽门螺杆感染相关的十二指肠溃疡-防止与幽门螺杆菌相关的消化性溃疡复发。
相比奥美拉唑,副作用更少,效果更佳,因此,该类药物临床应用中,埃索美拉唑镁的市场份额不断扩大。
本文参考相关文献,及国外药典涉及原料药及制剂标准,并优化改进,建立了埃索美拉唑镁稳定可靠的定量控制方法。
Agilent 1260型HPLC色谱仪,电子分析天平(梅特勒-托利多XS205型)。
[宝典]欧洲药典附录3.1.3.
![[宝典]欧洲药典附录3.1.3.](https://img.taocdn.com/s3/m/f3357cec760bf78a6529647d27284b73f2423679.png)
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3%–1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3%–二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
质量标准奥美拉唑

AS-NAI 质量标准
检查项目方法标准规定
性状目测
本品应为白色或类白色的冻干块状物或
粉末
鉴别一般鉴别试
验
本品显钠盐的鉴别反应(中国药典2010
年版二部附录Ⅲ)
HPLC
在对映体纯度测定项下记录的高效液相
色谱图中,供试品溶液主峰的保留时间
应与对照品溶液主峰的保留时间一致。
HPLC
在含量测定项下记录的高效液相色谱图
中,供试品溶液主峰的保留时间应与对
照品溶液主峰的保留时间一致。
溶液的颜色与澄清度溶液颜色检
查法
澄清度检查
法
取本品1瓶,加水1ml使溶解,溶液
应澄清无色,如显浑浊,与1号浊度标准
液(中国药典2010版二部附录IX B)比
较,不得更浓;如显色,与黄色1号标准
比色液(中国药典2010版二部附录IX A)
比较,不得更深。
碱度碱度检查法
取本品1瓶,加水1ml溶解后,依
法测定(中国药典2010版二部附录VI
H),pH值应为10.0~11.5
水分水分测定法
取本品适量,以无水甲醇为溶剂,照
水分测定法(中国药典2010年版二部附
录Ⅷ M第一法)测定,含水分不得过
0.5%。
有关物质HPLC 按面积归一化法计算,杂质C(碸)的量不得过0.2%、杂质A(咪唑), 杂质B (氮氧化物)和杂质D(硫醚)的量均不得过0.1%,其他单个杂质的量不得过0.1%,杂质总量不得过0.5%。
对映体纯度HPLC 纯度不低于99.5%
含量HPLC 含埃索美拉唑钠(C17H18N3NaO3S)应为标示量的90%~110%。
埃索美拉唑钠盐(原料药)标准 USP MC - Esomeprazole Sodium - 2012-07-23

Acceptance criteria of the impurity, in an appropriate diluent Sample solution: Esomeprazole Sodium in an appropriate diluent Analytical system: Use a procedure validated as described in MC general chapter Assessing Validation Parameters for Reference and Acceptable Procedures <10>.
ASSAY • Procedure
Solution A: 10 mM ammonium acetate in water. Adjust with ammonium hydroxide solution to a pH of 7.0. Solution B: Methanol and acetonitrile (1:1) Mobile phase: See Table 1.
IMPURITIES • Residue on Ignition <281>: NMT 0.1% • Elemental Impurities <232>: Proceed as directed in the chapter. • Residual Solvents <467>: Proceed as directed in the chapter. • Organic Impurities
埃索美拉唑镁usp34

© 2010 USPC Official 5/1/11 - 7/31/11 USP Monographs: Esomeprazole ...
页码,Байду номын сангаас/7
prepared atomic absorption standard solution. [NOTE—Store the solution in a plastic bottle. ] Standard solution A: Transfer 10.0 mL of Standard stock solution to a 500-mL volumetric flask, add 50 mL of 1 N hydrochloric acid, and dilute with water to volume. Transfer 20.0 mL of this solution to a 200-mL volumetric flask, and dilute with water to volume. [NOTE —This solution contains 2 µg/mL of magnesium. ] Standard solution B: Combine 5.0 mL of Standard solution A and 4.0 mL of Lanthanum solution , and dilute with water to 100.0 mL (0.1 µg/mL). Standard solution C: Combine 10.0 mL of Standard solution A and 4.0 mL of Lanthanum solution , and dilute with water to 100.0 mL (0.2 µg/mL). Standard solution D: Combine 15.0 mL of Standard solution A and 4.0 mL of Lanthanum solution , and dilute with water to 100.0 mL (0.3 µg/mL). Standard solution E: Combine 20.0 mL of Standard solution A and 4.0 mL of Lanthanum solution , and dilute with water to 100.0 mL (0.4 µg/mL). Standard solution F: Combine 25.0 mL of Standard solution A and 4.0 mL of Lanthanum solution , and dilute with water to 100.0 mL (0.5 µg/mL). [NOTE— Concentrations of the Standard solutions and the Sample solution may be modified to fit the linear or working range of the instrument. When using instruments with a linear calibration graph, the number of Standard solutions can be reduced. ] Blank solution: Transfer 4.0 mL of Lanthanum solution to a 100-mL volumetric flask, and dilute with water to volume. Sample solution: Transfer 250 mg of Esomeprazole Magnesium to a 100-mL volumetric flask, add 20 mL of 1 N hydrochloric acid, swirl until dissolved, and dilute with water to volume. Allow to stand for 30 min. Transfer 10.0 mL of this solution to a 200-mL volumetric flask, and dilute with water to volume. Transfer 10.0 mL of the solution to another 100 -mL volumetric flask, add 4.0 mL of Lanthanum solution , and dilute with water to volume. Spectrometric conditions (See Spectrophotometry and Light-Scattering 851 .) Mode: Atomic absorption spectrophotometer Flame: Air–acetylene Analytical wavelength: 285.2 nm Analysis Samples: Standard solution B, Standard solution C, Standard solution D, Standard solution E , Standard solution F, Blank solution , and Sample solution Determine the concentration, C s , in µg/mL, of magnesium in the Sample solution using the calibration graph. Calculate the percentage of magnesium in the portion of Esomeprazole Magnesium taken:
埃索美拉唑杂质
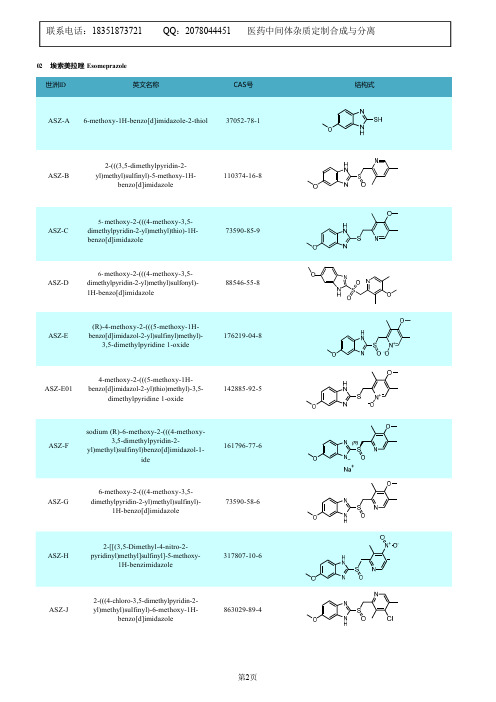
1227380-90-6
ASZ-H193
6-methoxy-2-(((4-methoxy-3,5dimethylpyridin-2-yl)methyl)sulfinyl)-1methyl-1H-benzo[d]imidazole AND 5methoxy-2-(((4-methoxy-3,5dimethylpyridin-2-yl)methyl)sulfinyl)-1methyl-1H-benzo[d]imidazole
73590-58-6
ASZ-H
2-[[(3,5-Dimethyl-4-nitro-2pyridinyl)methyl]sulfinyl]-5-methoxy1H-benzimidazole
317807-10-6
ASZ-J
2-(((4-chloro-3,5-dimethylpyridin-2yl)methyl)sulfinyl)-6-methoxy-1Hbenzo[d]imidazole
784143-42-6 89352-76-1
ASZ-H215/01
4sodium 2-(((3,5-dimethyl-4oxidopyridin-2-yl)methyl)sulfinyl)-5methoxybenzo[d]imidazol-1-ide
1803449-15-1 (free base: 301669-82-9)
863029-89-4
第2页
联系电话:18351873721
QQ:2078044451
医药中间体杂质定制合成与分离
ASZ-K
(((4-methoxy-3,5-dimethylpyridin-2yl)methyl)sulfinyl)-1Hbenzo[d]imidazole compound with 6- methoxy-1-((4-methoxy-3,5dimethylpyridin-2-yl)methyl)-2-(((4methoxy-3,5-dimethylpyridin-2yl)methyl)sulfinyl)-1Hbenzo[d]imidazole (1:1)
埃索美拉唑镁肠溶微丸的制备与质量控制

埃索美拉唑镁肠溶微丸的制备与质量控制摘要:目的:制备埃索美拉唑镁肠溶微丸,建立质量控制方案。
方法:应用流化床包衣法(底喷),成功制备出埃索美拉唑镁肠溶微丸,进行溶出度、耐酸力测定,评价埃索美拉唑镁肠溶微丸的质量。
结果:pH值6.8的人工肠液中,自制埃索美拉唑镁肠溶微丸的溶出度>86%,pH值1.2的氯化氢溶液中(模拟胃液环境),自制埃索美拉唑镁肠溶微丸的累积溶出度<2%。
结论:人工肠液中,自制埃索美拉唑镁肠溶微丸的溶出较完全,人工胃液中,自制埃索美拉唑镁肠溶微丸的耐酸力较好,该制备工艺可靠,药物质量理想,可推广。
关键词:埃索美拉唑镁肠溶微丸;制备工艺;质量控制埃索美拉唑镁主要应用于胃食管反流性疾病的临床治疗中,但容易被酸性物质所催化降解,为避免其被胃酸破坏、降低疗效,需要进行包衣处理,也就是制备埃索美拉唑镁肠溶微丸。
1.材料与方法1.1材料与仪器1)材料:埃索美拉唑镁三水合物、滑石粉、空白糖丸、羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、柠檬酸三乙酯、乙腈。
2)仪器:小型流化床包衣机、磁力搅拌器、恒流泵、智能溶出仪、分析天平、高效液相色谱仪。
1.2埃索美拉唑镁肠溶微丸的制备工艺1)载药微丸的制备。
以3% HPMC水溶液为黏合剂,并加入埃索美拉唑镁,从而制备为混悬液。
在小型流化床包衣机中对空白糖丸进行加工,应用底喷工艺,将其制成载药丸心,工艺参数:雾化压力0.1兆帕至0.11兆帕、床温30±1摄氏度、平衡状态下的喷液流速每分钟1.5毫升。
载药丸心制备完成后,流化干燥30分钟。
2)隔离微丸的制备。
取处方量的HPC、滑石粉溶于水中,制备隔离包衣液,浓度为3.5%。
在小型流化床包衣机中应用底喷工艺进行包衣处理,工艺参数:雾化压力0.1兆帕、床温37摄氏度、喷液流速每分钟3.5毫升。
包衣完成后,流化干燥30分钟。
载药微丸包衣的目的是,由于埃索美拉唑镁在酸性环境下容易发生分解,因此需要将埃索美拉唑镁制备为肠溶剂,但使用的是弱酸性的肠溶包衣材料,所以应进行隔离包衣处理,避免肠溶材料与埃索美拉唑镁发生化学反应[1]。
埃索美拉唑镁标准-EP7.0

CHARACTERS Appearance: white or slightly coloured powder, slightly hygroscopic. Solubility : slightly soluble in water, soluble in methanol, practically insoluble in heptane.
ห้องสมุดไป่ตู้
TESTS
A. pXe=ntNa-nCoHl,3: 1-[(2-chlorophenyl)(methylimino)methyl]cycloC. X = O : (2-chlorophenyl)(1-hydroxycyclopentyl)methanone,
B. (2RS)-2-(2-chlorophenyl)-2-hydroxycyclohexanone,
identification CRS and the chromatogram obtained with reference solution (b) to identify the peak due to impurity E ; — use the chromatogram obtained with reference solution (a) to identify the peak due to impurity D. Relative retention with reference to esomeprazole (retention time = about 9 min) : impurity E = about 0.6 ; impurity D = about 0.8. System suitability : reference solution (a) : — resolution : minimum 3.0 between the peaks due to impurity D and omeprazole. If necessary, adjust the pH of the aqueous part of the mobile phase or its proportion of acetonitrile ; an increase in the pH will improve the resolution. Limits : — impurity D : maximum 0.2 per cent ; — impurity E : maximum 0.1 per cent; — unspecified impurities : for each impurity, maximum 0.10 per cent ; — total : maximum 0.5 per cent ; — disregard limit : 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (c) (0.05 per cent).