生物信息学研究方法
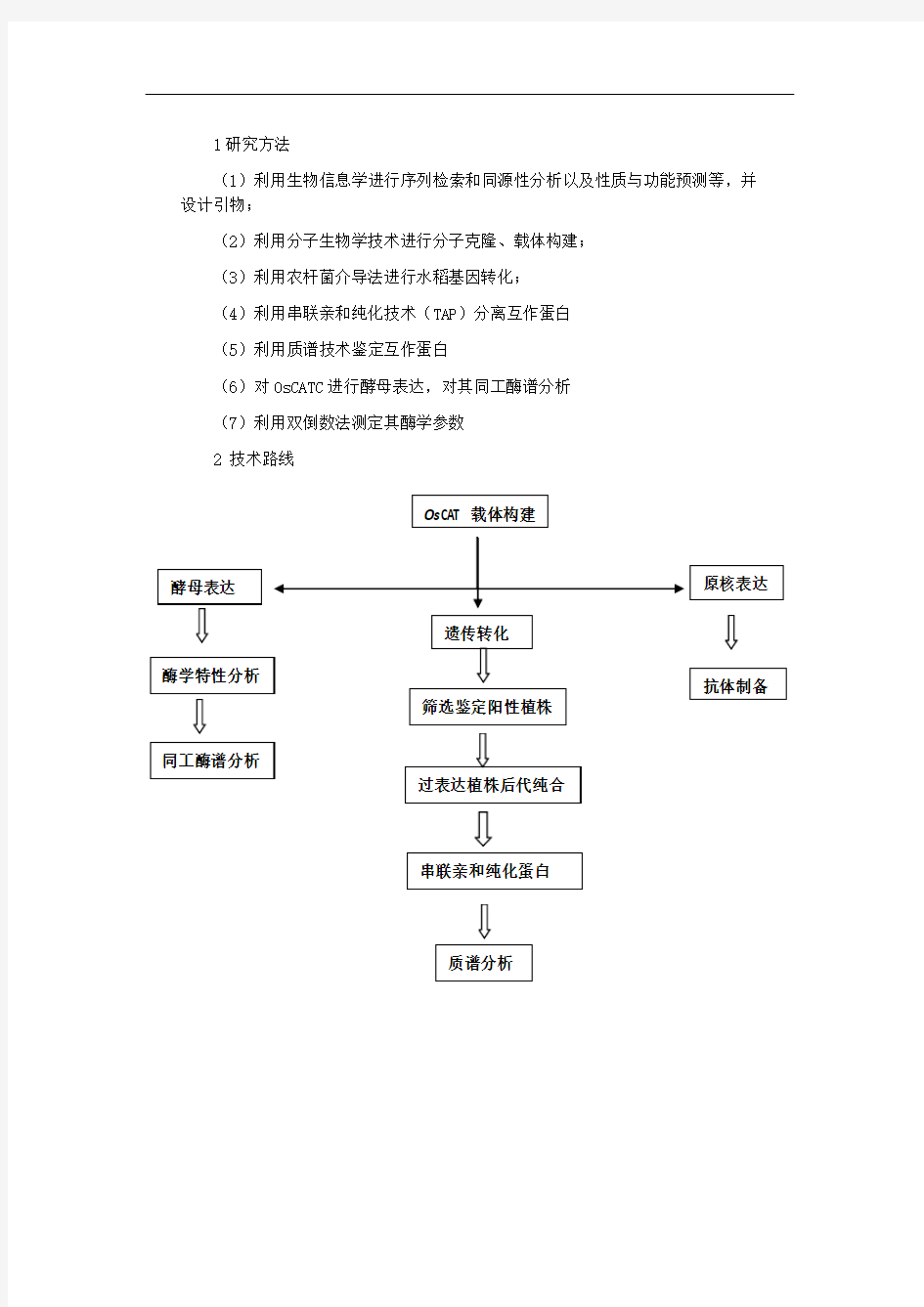
1研究方法
(1)利用生物信息学进行序列检索和同源性分析以及性质与功能预测等,并设计引物;
(2)利用分子生物学技术进行分子克隆、载体构建;
(3)利用农杆菌介导法进行水稻基因转化;
(4)利用串联亲和纯化技术(TAP )分离互作蛋白
(5)利用质谱技术鉴定互作蛋白
(6)对OsCATC 进行酵母表达,对其同工酶谱分析
(7)利用双倒数法测定其酶学参数
2 技术路线
高通量测序生物信息学分析(内部极品资料,初学者必看)
基因组测序基础知识 ㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。 目前国际上通用的基因组De Novo测序方法有三种: 1. 用Illumina Solexa GA IIx 测序仪直接测序; 2. 用Roche GS FLX Titanium直接完成全基因组测序; 3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx 进行深度测序,完成基因组拼接。 采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。 实验流程: 公司服务内容 1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头, 去污染);序列组装达到精细图标准 2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展 示平台搭建 1.基因组De Novo测序对DNA样品有什么要求?
(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。 (2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (4) 基因组De Novo组装完毕后需要构建BAC或Fosmid文库进行测序验证,用于BAC 或Fosmid文库构建的样品需要保证跟De Novo测序样本同一来源。 2. De Novo有几种测序方式 目前3种测序技术 Roche 454,Solexa和ABI SOLID均有单端测序和双端测序两种方式。在基因组De Novo测序过程中,Roche 454的单端测序读长可以达到400 bp,经常用于基因组骨架的组装,而Solexa和ABI SOLID双端测序可以用于组装scaffolds和填补gap。下面以solexa 为例,对单端测序(Single-read)和双端测序(Paired-end和Mate-pair)进行介绍。Single-read、Paired-end和Mate-pair主要区别在测序文库的构建方法上。 单端测序(Single-read)首先将DNA样本进行片段化处理形成200-500bp的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flow cell上生成DNA簇,上机测序单端读取序列(图1)。 Paired-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paired-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序(图2)。 图1 Single-read文库构建方法图2 Paired-end文库构建方法
生物信息学分析实践
水稻瘤矮病毒(RGDV)外层衣壳蛋白 P8的同源模建 高芳銮(Raindy) 同源模建(homology modeling) ,也叫比较模建(Compatative modeling),其前提是一个或多个同源蛋白质的结构已知,当两个蛋白质的序列同源性高于35%,一般情况下认为它们的三维结构基本相同;序列同源性低于30%的蛋白质难以得到理想的结构模型。同源模建是目前最为成功且实用的蛋白质结构预测方法, SWISS-MODEL 是由SwissProt 提供的目前最著名的蛋白质三级结构预测服务器,创建于1993年,面向全世界的生物化学与分子生物学研究工作者提供免费的自动模建服务。SWISS-MODEL 服务器提供的同源模建有两种工作模式:首选模式(First Approach mode)和 项目模式(Project mode)。 本实例以RGDV P8蛋白为研究对象采用首选模式进行同源模建。 图1 SWISS-MODEL 的主界面 操作流程如下: 1.选择模式 单击左侧的“MENU ”菜单下方的“First Approach mode ”,右侧窗口自动SWISS-MODEL 工作窗口,在相应文本框中分别输入的E-mail 、项目标题、待模建的蛋白质序列,SWISS-MODEL 支持以FASTA 格式直接输入或提交UniProt 的登录号,如图2所示。 《生物信息学分析实践》样 稿
图2 SWISS-MODEL 的序列提交页面 2.参数设置 当前版本只有一个选项可设置,如果用户需要使用指定的模板,可在“Use a specific template ”后的输入框填入ExPDB 晶体图像数据库中的模板代码,其格式为“PDBCODE+ChainID ”,如“1uf2P ”。本例不使用指定模板,默认留空。完毕,点击“Submit Modeling Request ”提交模建请求,服务器返回提交成功的提示,如图3所示: 图3 成功提交 SWISS-MODEL WORKSPACEW 页面会自动刷新,直至模建完成,如图4所示,同时模建结果也会发送到指定的邮箱。 3结果解读 点击下图右上方的“Print/Save this page as ”后的图标,可以将整个结果以PDF 文档格式保存到本地计算机中。模建结果给出了五个部分的信息:模建详情(Model Details)、比对信息(Alignment)、模建评价 (Anolea/Gromos/Verify3D)、模建日志(Modelling log)、模板选择日志(Template Selection Log)。 《生物信息学分析实践》样稿
生物信息学分析
4、生物信息学分析 通过核苷酸序列数据库和基因序列同源性在线分析途径初步对Rv2029c基因进行分类整理。由于结核分枝杆菌耐利福平野生株与核苷酸序列数据库KEGG GENES中的结核分枝杆菌标准株H37Rv的匹配率为100%,以下对基因的分析按照结核分枝杆菌标准株H37Rv的数据库信息进行,即完全匹配的1020bp长度序列(本次提取基因中包含上下游引物等序列,较长,1346bp)。 4.1基本信息 表1 基因基本信息 4.2基因组信息 表2 基因组信息
5、PLN02341(PfkB型碳水化合物激酶家族蛋白),位点208-294 6、PTZ0029(核糖激酶),位点205-301 药物靶点1、同源基因没有药物靶点 2、非同源但序列相似基因没有药物靶点 图3 蛋白结构域 4.3蛋白表达 4.3.1 二级结构分析 预测结果显示,PfkB蛋白的二级结构中β转角占46.61%,α螺旋占33.63%,β折叠占19.76%。转角结构和螺旋结构构成了结核分枝杆菌PfkB蛋白二级结构的骨架。
图4 蛋白二级结构 4.3.2 跨膜区分析 Tuberculist跨膜蛋白预测结果表明:蛋白长度339aa,预测跨膜蛋白数0。 图5 蛋白跨膜区分析 4.3.3 信号肽预测 Predict Protein分析表明PfkB蛋白氨基酸残基没有信号肽,由此推断此蛋白不包含信号肽,不是分泌型蛋白质。
图6 蛋白信号肽预测 4.3.4 疏水性分析 分析结果显示,蛋白最大疏水指数为2.411,最小疏水指数为-2.372。
图7 蛋白疏水性分析 4.3.5 DNA同源性分析 表3 基因同源性分析 菌株序列覆盖 率 E值一致性 Mycobacterium tuberculosis strain Beijing-like, complete genome 100% 0.0 100% Mycobacterium bovis subsp. bovis AF2122/97 complete genome 100% 0.0 100% Mycobacterium tuberculosis 18b genome 100% 0.0 100% Mycobacterium tuberculosis H37RvSiena, complete genome 100% 0.0 100% Mycobacterium tuberculosis str. Kurono DNA, complete genome 100% 0.0 100% Mycobacterium tuberculosis 49-02 complete 100% 0.0 100%
生物信息学的主要研究内容
常用数据库 在DNA序列方面有GenBank、EMBL和等 在蛋白质一级结构方面有SWISS-PROT、PIR和MIPS等 在蛋白质和其它生物大分子的结构方面有PDB等 在蛋白质结构分类方面有SCOP和CATH等 生物信息学的主要研究内容 1、序列比对(Alignment) 基本问题是比较两个或两个以上符号序列的相似性或不相似性。序列比对是生物信息学的基础,非常重要。两个序列的比对有较成熟的动态规划算法,以及在此基础上编写的比对软件包BLAST和FASTA,可以免费下载使用。这些软件在数据库查询和搜索中有重要的应用。 2、结构比对 基本问题是比较两个或两个以上蛋白质分子空间结构的相似性或不相似性。已有一些算法。 3、蛋白质结构预测,包括2级和3级结构预测,是最重要的课题之一 从方法上来看有演绎法和归纳法两种途径。前者主要是从一些基本原理或假设出发来预测和研究蛋白质的结构和折叠过程。分子力学和分子动力学属这一范畴。后者主要是从观察和总结已知结构的蛋白质结构规律出发来预测未知蛋白质的结构。同源模建(Homology)和指认(Threading)方法属于这一范畴。虽然经过30余年的努力,蛋白结构预测研究现状远远不能满足实际需要。 4、计算机辅助基因识别(仅指蛋白质编码基因)。最重要的课题之一 基本问题是给定基因组序列后,正确识别基因的范围和在基因组序列中的精确位置.这是最重要的课题之一,而且越来越重要。经过20余年的努力,提出了数十种算法,有十种左右重要的算法和相应软件上网提供免费服务。原核生物计算机辅助基因识别相对容易些,结果好一些。从具有较多内含子的真核生物基因组序列中正确识别出起始密码子、剪切位点和终止密码子,是个相当困难的问题,研究现状不能令人满意,仍有大量的工作要做。 5、非编码区分析和DNA语言研究,是最重要的课题之一 在人类基因组中,编码部分进展总序列的3~5%,其它通常称为“垃圾”DNA,其实一点也不是垃圾,只是我们暂时还不知道其重要的功能。分析非编码区DNA 序列需要大胆的想象和崭新的研究思路和方法。DNA序列作为一种遗传语言,不仅体现在编码序列之中,而且隐含在非编码序列之中。 6、分子进化和比较基因组学,是最重要的课题之一 早期的工作主要是利用不同物种中同一种基因序列的异同来研究生物的进化,构建进化树。既可以用DNA序列也可以用其编码的氨基酸序列来做,甚至于可通过相关蛋白质的结构比对来研究分子进化。以上研究已经积累了大量的工作。近年来由于较多模式生物基因组测序任务的完成,为从整个基因组的角度来研究分子进化提供了条件。 7、序列重叠群(Contigs)装配 一般来说,根据现行的测序技术,每次反应只能测出500或更多一些碱基对的序列,这就有一个把大量的较短的序列全体构成了重叠群(Contigs)。逐步把它们拼接起来形成序列更长的重叠群,直至得到完整序列的过程称为重叠群装配。拼接EST数据以发现全长新基因也有类似的问题。已经证明,这是一个NP-完备
生物信息学复习资料全
一、名词解释(31个) 1.生物信息学:广义:应用信息科学的方法和技术,研究生物体系和生物过程 息的存贮、信息的涵和信息的传递,研究和分析生物体细胞、组织、器官的生理、病理、药理过程中的各种生物信息,或者也可以说成是生命科学中的信息科学。狭义:应用信息科学的理论、方法和技术,管理、分析和利用生物分子数据。 2.二级数据库:对原始生物分子数据进行整理、分类的结果,是在一级数据库、 实验数据和理论分析的基础上针对特定的应用目标而建立的。 3.多序列比对:研究的是多个序列的共性。序列的多重比对可用来搜索基因组 序列的功能区域,也可用于研究一组蛋白质之间的进化关系。 4.系统发育分析:是研究物种进化和系统分类的一种方法,其常用一种类似树 状分支的图形来概括各种(类)生物之间的亲缘关系,这种树状分支的图形称为系统发育树。 5.直系同源:如果由于进化压力来维持特定模体的话,模体中的组成蛋白应该 是进化保守的并且在其他物种中具有直系同源性。 指的是不同物种之间的同源性,例如蛋白质的同源性,DNA序列的同源性。(来自百度) 6.旁系(并系)同源:是那些在一定物种中的来源于基因复制的蛋白,可能会 进化出新的与原来有关的功能。用来描述在同一物种由于基因复制而分离的同源基因。(来自百度) 7.FASTA序列格式:将一个DNA或者蛋白质序列表示为一个带有一些标记的 核苷酸或氨基酸字符串。 8.开放阅读框(ORF):是结构基因的正常核苷酸序列,从起始密码子到终止 密码子的阅读框可编码完整的多肽链,其间不存在使翻译中断的终止密码子。(来自百度) 9.结构域:大分子蛋白质的三级结构常可分割成一个或数个球状或纤维状的区 域,折叠得较为紧密,各行其功能,称为结构域。 10.空位罚分:序列比对分析时为了反映核酸或氨基酸的插入或缺失等而插入空 位并进行罚分,以控制空位插入的合理性。(来自百度) 11.表达序列标签:通过从cDNA文库中随机挑选的克隆进行测序所获得的部分 cDNA的3’或5’端序列。(来自文献) 12.Gene Ontology 协会: 13.HMM 隐马尔可夫模型:将核苷酸序列看成一个随机序列,DNA序列的编 码部分与非编码部分在核苷酸的选用频率上对应着不同的Markov模型。14.一级数据库:数据库中的数据直接来源于实验获得的原始数据,只经过简单 的归类整理和注释 15.序列一致性:指同源DNA顺序的同一碱基位置的相同的碱基成员, 或者蛋 白质的同一氨基酸位置的相同的氨基酸成员, 可用百分比表示。 16.序列相似性:指同源蛋白质的氨基酸序列中一致性氨基酸和可取代氨基酸所 占的比例。 17.Blastn:是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将 同所查序列作一对一地核酸序列比对。(来自百度) 18.Blastp:是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐 一地同每条所查序列作一对一的序列比对。(来自百度)
基因组学与生物信息学教案
《基因组学与生物信息学》教案 授课专业:生物学大类各专业 课程名称:基因组学与生物信息学 主讲教师:夏庆友程道军赵萍徐汉福
课程说明 一、课程名称:基因组学与生物信息学 二、总课时数:36学时(理论27学时实验9学时) 三、先修课程:遗传学、分子生物学、基因工程 四、使用教材: 杨金水. 基因组学. 北京:高等教育出版社,2002. 张成岗. 贺福初, 生物信息学方法与实践. 北京:科学出版社,2002. 五、教学参考书: T.A.布朗著,袁建刚译著,基因组(2rd版),北京:科学出版社,2006. 沈桂芳,丁仁瑞,走向后基因组时代的分子生物学,杭州:浙江教育出版社,2005. 罗静初译,生物信息学概论,北京:北京大学出版社,2002. 六、考核方式:考查 七、教案编写说明: 教案又称课时授课计划,是任课教师的教学实施方案。任课教师应遵循专业教学计划制订的培养目标,以教学大纲为依据,在熟悉教材、了解学生的基础上,结合教学实践经验,提前编写设计好每门课程每个章、节或主题的全部教学活动。教案可以按每堂课(指同一主题连续1~2节课)设计编写。教案编写说明如下: 1、编号:按施教的顺序标明序号。 2、教学课型表示所授课程的类型,请在相应课型栏内选择打“√”。 3、题目:标明章、节或主题。 4、教学内容:是授课的核心。将授课的内容按逻辑层次,有序设计编排,必要时标以“*”、“#”“?” 符号分别表示重点、难点或疑点。 5、教学方式既教学方法,如讲授、讨论、示教、指导等。教学手段指教科书、板书、多媒体、模型、 标本、挂图、音像等教学工具。 6、讨论、思考题和作业:提出若干问题以供讨论,或作为课后复习时思考,亦可要求学生作为作业 来完成,以供考核之用。 7、参考书目:列出参考书籍、有关资料。 8、日期的填写系指本堂课授课的时间。
生物信息学分析实验报告
1、分别写出2010年以来,国际上与Ovarian cancer、Breast cancer、Leukemia相关的文献有多少篇?写出3篇研究性论文标题和摘要,写出5篇综述性论文标题和摘要; 数据库:科学引文索引数据库(SCI:Science Citation Index) https://www.360docs.net/doc/8918082566.html, 与Ovarian cancer相关的文献有11,303篇 与Breast cancer相关的文献有56,209篇 与Leukemia相关的文献有32,912篇 综述性论文标题和摘要 1.Hemochromatosis and ovarian cancer 摘要:Evaluation of: Gannon PO, Medelci S, Le Page C et al. Impact of hemochromatosis gene (HFE) mutations on epithelial ovarian cancer risk and prognosis. Int. J. Cancer 128(10), 2326-2334 (2011). The frequency of two mutations (C282Y and D62H) of the hemochromatosis gene were investigated in women with ovarian cancer. A single allele mutation of the C282Y but not the H63D gene product was detected in 8-9% of women with benign ovarian tumors (n = 124) and ovarian cancers (n = 360) compared with 2.5% for controls (n = 80) representing a 4.9-fold increase in risk. With high-grade serous ovarian cancers (n = 179), the survival rate of women with a single allele C282Y mutation was reduced from 39 to 19 months. These results implicate mutations of the hemochromatosis gene in the generation and severity of ovarian cancers, which may have prognostic value. 2.Differences between women who pursued genetic testing for hereditary breast and ovarian cancer and their at-risk relatives who did not. 摘要: Purpose/Objectives: To (a) examine differences in appraisals of hereditary breast and ovarian cancer (HBOC), psychological distress, family environment, and decisional conflict between women who pursued genetic testing and their at-risk relatives who did not, and (b) examine correlations among appraisals of HBOC, psychological distress, family environment, and decisional conflict regarding genetic testing in these two cohorts of women.Design: Descriptive, cross-sectional cohort study.Setting: Two clinics affiliated with a major research university in the midwestern United States.Sample: 372 women aged 18 years and older. 200 pursued genetic testing for BRCA1 and BRCA2 mutations (probands) and 172 of their female relatives who had a greater than 10% prior probability of being a mutation carrier but had not pursued testing.Methods: After providing informed consent, probands and relatives were mailed self-administered questionnaires.Main Research Variables: Perceived risk, knowledge of HBOC risk factors and modes of gene inheritance, perceived severity, perceived controllability, psychological distress, family relationships, family communication, and decisional conflict about genetic testing.Findings: T tests revealed that probands perceived higher risk and had more psychological distress associated with breast cancer. Probands had more knowledge regarding risk factors and gene inheritance, and greater decisional conflict regarding genetic testing. Relatives reported higher perceived severity and controllability. No differences were observed in family relationships and family communication between probands
蛋白质组学生物信息学分析介绍
生物信息学分析FAQ CHAPTER ONE ABOUT GENE ONTOLOGY ANNOTATION (3) 什么是GO? (3) GO和KEGG注释之前,为什么要先进行序列比对(BLAST)? (3) GO注释的意义? (3) GO和GOslim的区别 (4) 为什么有些蛋白没有GO注释信息? (4) 为什么GO Level 2的统计饼图里蛋白数目和差异蛋白总数不一致? (4) 什么是差异蛋白的功能富集分析&WHY? (4) GO注释结果文件解析 (5) Sheet TopBlastHits (5) Sheet protein2GO/protein2GOslim (5) Sheet BP/MF/CC (6) Sheet Level2_BP/Level2_MF/Level2_CC (6) CHAPTER TWO ABOUT KEGG PATHWAY ANNOTATION (7) WHY KEGG pathway annotation? (7) KEGG通路注释的方法&流程? (7) KEGG通路注释的意义? (7) 为什么有些蛋白没有KEGG通路注释信息? (8) 什么是差异蛋白的通路富集分析&WHY? (8) KEGG注释结果文件解析 (8) Sheet query2map (8) Sheet map2query (9) Sheet TopMapStat (9) CHAPTER THREE ABOUT FEATURE SELECTION & CLUSTERING (10) WHY Feature Selection? (10)
聚类分析(Clustering) (10) 聚类结果文件解析 (10) CHAPTER FOUR ABOUT PROTEIN-PROTEIN INTERACTION NETWORK (12) 蛋白质相互作用网络分析的意义 (12) 蛋白质相互作用 VS生物学通路? (12) 蛋白质相互作用网络分析结果文件解析 (12)
生物信息学中的机器学习方法
生物信息学中的机器学习方法 摘要:生物信息学是一门交叉学科,包含了生物信息的获取、管理、分析、解释和应用等方面,兴起于人类基因组计划。随着人类基因组计划的完成与深入,生物信息的研究工作由原来的计算生物学时代进入后基因组时代,后基因组时代中一个最重要的分支就是系统生物学。本文从信息科学的视角出发,详细论述了机器学习方法在计算生物学和系统生物学中的若干应用。 关键词:生物信息学;机器学习;序列比对;人类基因组;生物芯片 1.相关知识 1.1 生物信息学 生物信息学时生物学与计算机科学以及应用数学等学科相互交叉而形成的一门新兴学科。它综合运用生物学、计算机科学和数学等多方面知识与方法,来阐明和理解大量生物数据所包含的生物学意义,并应用于解决生命科学研究和生物技术相关产业中的各种问题。 生物信息学主要有三个组成部分:建立可以存放和管理大量生物信息学数据的数据库;研究开发可用于有效分析与挖掘生物学数据的方法、算法和软件工具;使用这些工具去分析和解释不同类型的生物学数据,包括DNA、RNA和蛋白质序列、蛋白质结构、基因表达以及生化途径等。 生物信息学这个术语从20世纪90年代开始使用,最初主要指的是DNA、RNA及蛋白质序列的数据管理和分析。自从20世纪60年代就有了序列分析的计算机工具,但是那时并未引起人们很大的关注,直到测序技术的发展使GenBank之类的数据库中存放的序列数量出现了迅猛的增长。现在该术语已扩展到几乎覆盖各种类型的生物学数据,如蛋白质结构、基因表达和蛋白质互作等。 目前的生物信息学研究,已从早期以数据库的建立和DNA序列分析为主的阶段,转移到后基因组学时代以比较基因组学(comparative genomics)、功能基因组学(functional genomics)和整合基因组学(integrative genomics)为中心的新阶段。生物信息学的研究领域也迅速扩大。生物信息学涉及生物学、计算机学、数学、统计学等多门学科,从事生物信息学研究的工作者或生物信息学家可以来自以上任何一个领域而侧重于生物信息学的不同方面。事实上,我们今天正需要具备各种背景知识、才能和研究思路的研究人员,集思广益
浅谈生物信息学在生物方面的应用
浅谈生物信息学在生物方面的应用 生物信息学(bioinformaLics)是以核酸和蛋白质等生物大分子数据库及其相关的图书、文献、资料为主要对象,以数学、信息学、计算机科学为主要手段,对浩如烟海的原始数据和原始资料进行存储、管理、注释、加工,使之成为具有明确生物意义的生物信息。并通过对生物信息的查询、搜索、比较、分析,从中获得基因的编码、凋控、遗传、突变等知识;研究核酸和蛋白质等生物大分子的结构、功能及其相互关系;研究它们在生物体内的物质代谢、能量转移、信息传导等生命活动中的作用机制。 从生物信息学研究的具体内容上看,生物信息学可以用于序列分类、相似性搜索、DNA 序列编码区识别、分子结构与功能预测、进化过程的构建等方面的计算工具已成为变态反应研究工作的重要组成部分。针对核酸序列的分析就是在核酸序列中寻找过敏原基因,找出基因的位置和功能位点的位置,以及标记已知的序列模式等过程。针对蛋白质序列的分析,可以预测出蛋白质的许多物理特性,包括等电点分子量、酶切特性、疏水性、电荷分布等以及蛋白质二级结构预测,三维结构预测等。 生物信息学中的主要方法有:序列比对,结构比对,蛋白质结构的预测,构造分子进化树,聚类等。基因芯片是基因表达谱数据的重要来源。目前生物信息学在基因芯片中的应用主要体现在三个方面。 1、确定芯片检测目标。利用生物信息学方法,查询生物分子信息数据库,取得相应的序列数据,通过序列比对,找出特征序列,作为芯片设计的参照序列。 2、芯片设计。主要包括两个方面,即探针的设计和探针在芯片上的布局,必须根据具体的芯片功能、芯片制备技术采用不同的设计方法。 3、实验数据管理与分析。对基因芯片杂交图像处理,给出实验结果,并运用生物信息学方法对实验进行可靠性分析,得到基因序列变异结果或基因表达分析结果。尽可能将实验结果及分析结果存放在数据库中,将基因芯片数据与公共数据库进行链接,利用数据挖掘方法,揭示各种数据之间的关系。 生物信息学在人类基因组计划中也具有重要的作用。 大规模测序是基因组研究的最基本任务,它的每一个环节都与信息分析紧密相关。目前,从测序仪的光密度采样与分析、碱基读出、载体标识与去除、拼接与组装、填补序列间隙,到重复序列标识、读框预测和基因标注的每一步都是紧密依赖基因组信息学的软件和数据库的。特别是拼接和填补序列间隙更需要把实验设计和信息分析时刻联系在一起.拼接与组装中的难点是处理重复序列,这在含有约30%重复序列的人类基因组中显得尤其突出。 人类基因组的工作草图即将完成,因此发现新基因就成了当务之急。使用基因组信息学的方法通过超大规模计算是发现新基因的重要手段,可以说大部分新基因是靠理论方法预测出来的。比如啤酒酵母完整基因组(约1300万bp)所包含6千多个基因,大约60%是通过信息分析得到的。 当人类基因找到之后,自然要解决的问题是:不同人种间基因有什么差别;正常人和病人基因又有什么差别。”这就是通常所说的SNPs(单核苷酸多态性)。构建SNPs及其相关数据库是基因组研究走向应用的重要步骤。1998年国际已开展了以EST为主发现新Spps 的研究。在我国开展中华民族SNPs研究也是至重要的。总之,生物信息学不仅将赋予人们各种基础研究的重要成果,也会带来巨大的经济效益和社会效益。在未来的几年中DNA 序列数据将以意想不到的速度增长,这更离不开利用生物信息学进行各类数据的分析和解释,研制有效利用和管理数据新工具。生物信息学在功能基因组学同样具有重要的应用目前应用最多的是同源序列比较、模式识别以及蛋白结构预测。所谓同源序列,是指从某一共同祖先经趋异进化而形成的不同序列。利用数据库搜索找出未知核酸或蛋白的同源序列,是序列分析的基础[lol。如利用BLASTn和BLASTx两种软件分别进行核苷酸和氨基
生物信息学分析
生物信息学分析 生物信息学难吗? 经常有人向我问这个问题,这有什么疑问吗?如果不难学,根本就不用问我这个问题。也无需投入那么多时间精力就能掌握,更无需花费三四千元参加线下的培训班,也不会月薪过万。所以,答案很肯定,道理很简单:生物信息比较难学。 为什么难学? 我总结里几点原因。首先,这是一个交叉学科,要求你既要有生物学的基础,又要有很强的计算机操作技能。这个就有点困难了。因为只是一个生物学就包括多个门类,有很多东西需要去学习,还需要学习计算机知识。很多人一门内容还没学明白,现在还得在加一门,这就属于祸不单行,雪上加霜,屋漏偏逢连夜雨。因此,这种既懂生物学,又懂计算机的复合型人才就比较短缺。而且,生物信息本质上属于数据挖掘,除了生物,计算机,到后面还需要极强的统计学知识才能做好数据分析,所以,还得加上统计学,也就是生物信息学=生物学+计算机科学+统计学三门学科的知识,这也就是为什么生物信息学比较难学。 第二个原因,生物信息本身就包括很多内容,比如DNA的分析,RNA的分析,甲基化的分析,蛋白质的分析等方面,每一
门类又完全不同,从物种方面来分,动物,植物,微生物,医学等有差别很大,很难有一劳永逸,放之四海而皆准的分析方法。 第三个原因就是生物信息是一门快速发展的学习,会出现很多新的测序方法,比如sanger测序,illumina,BGIseq,PacBio,IonTorrent,Nanopore等,每一个平台技术原理完全不同,因此数据特点也完全不同,这就需要针对每一个平台的数据做专门的学习,而且每个平台又在不断的推陈出现,可能今天你刚开发好的方法,产品升级了,都得推倒重来。还有很多新的技术,例如现在比较火的单细胞测序,Hi-C测序,Bionano测序等等内容,以后还出现更多新技术新方法,足够让你活到老,学到老。当然,你先要能活到老,吾生也有涯,而知也无涯。以有涯随无涯,殆已! 高风险才有高收益 当然啦,虽然你已经看到学习生物信息肯定是不容易了,门槛很高,但是呢,门槛高也有很多好处,就是挡住了一部分人,当你学会了,迈过门槛,你的身价就提高了。如果人人都很容易掌握了,那么也就不值钱了。所以,生物信息,前途是光明的,道路是曲折的。
生物信息学实验报告3(三)蛋白质序列分析
(三)蛋白质序列分析 实验目的:掌握蛋白质序列检索的操作方法,熟悉蛋白质基本性质分析,了解蛋白质结构分析和预测。 实验内容: 1、检索SOX-21蛋白质序列,利用ProParam工具进行蛋白质的氨基酸组成、分子质量、等电点、氨基酸组成、原子总数及疏水性(ProtScale工具)等理化性质的分析。 2、利用PredictProtein、PROF、HNN等软件预测分析蛋白质的二级结构;利用Scan Prosite软件对蛋白质进行结构域分析。 3、利用TMHMM、TMPRED、SOSUI等工具对蛋白质进行跨膜分析;采用PredictNLS进行核定位信号分析;利用PSORT进行蛋白质的亚细胞定位预测;利用CBS(http://www.cbs.dtu.dk/services/ProtFun/)网站工具预测蛋白的功能,将序列用Blocks、SMART、InterProScan、PFSCAN等搜索其保守序列的特征,进行motif 的结构分析。 4、利用Swiss-Model数据库软件预测该蛋白的三级结构,结果用蛋白质三维图象软件Jmol查看。CPHmodels 也是利用神经网络进行同源模建预测蛋白质结构的方法和网络服务器I-TASSER预测所选蛋白质的空间结构。 5、分析蛋白质的翻译后修饰:分析信号肽及其剪切位点: SignalIP http://www.cbs.dtu.dk/services/SignalP/;分析糖链连接点:分析O-连接糖蛋白, NetOGlyc,http://www.cbs.dtu.dk/services/NetOGlyc/;分析N-连接糖蛋白,NetNGlyc,http://www.cbs.dtu.dk/services/NetNGlyc/。 6、利用检索的序列,进行同源比对,获得并分析比对结果。 实验步骤 (一) 1、在NCBI 蛋白质数据库中查找SOX-21蛋白质序列分别选择爪蟾(Xenopus laevis)、小家鼠[Mus musculus]、猕猴[Macaca mulatt a]的SOX-21蛋白质序列,并保存其FASTA格式。 2、利用ProParam工具对SOX-21蛋白质序列进行理化性质的分子。 3、利用PredictProtein、PROF、HNN等软件预测分析蛋白质的二级结构;利用Scan Prosite软件对蛋白质进行结构域分析。 4、利用TMHMM、TMPRED、SOSUI等工具对蛋白质进行跨膜分析;采用
生物信息学完整版
一、名词解释 1. 生物信息学: 1)生物信息学包含了生物信息的获取、处理、分析、和解释等在内的一门交叉学科; 2)它综合运用了数学、计算机学和生物学的各种工具来进行研究; 3)目的在于阐明大量生物学数据所包含的生物学意义。 2. BLAST(Basic Local Alignment Search Tool) 直译:基本局部排比搜索工具 意译:基于局部序列排比的常用数据库搜索工具 含义:蛋白质和核酸序列数据库搜索软件系统及相关数据库 3. PSI-BLAST:是一种迭代的搜索方法,可以提高BLAST和FASTA的相似序列发现率。 4. 一致序列:这些序列是指把多序列联配的信息压缩至单条序列,主要的缺点是除了在特 定位置最常见的残基之外,它们不能表示任何概率信息。 5. HMM 隐马尔可夫模型:一种统计模型,它考虑有关匹配、错配和间隔的所有可能的组合 来生成一组序列排列。(课件定义)是蛋白质结构域家族序列的一种严格的统计模型,包括序列的匹配,插入和缺失状态,并根据每种状态的概率分布和状态间的相互转换来生成蛋白质序列。 6. 信息位点:由位点产生的突变数目把其中的一课树与其他树区分开的位点。 7. 非信息位点:对于最大简约法来说没有意义的点。 8. 标度树:分支长度与相邻节点对的差异程度成正比的树。 9. 非标度树:只表示亲缘关系无差异程度信息。 10. 有根树:单一的节点能指派为共同的祖先,从祖先节点只有唯一的路径历经进化到达其 他任何节点。 11. 无根树:只表明节点间的关系,无进化发生方向的信息,通过引入外群或外部参考物种, 可以在无根树中指派根节点。 12. 注释:指从原始序列数据中获得有用的生物学信息。这主要是指在基因组DNA中寻找基 因和其他功能元件(结构注释),并给出这些序列的功能(功能注释)。 13. 聚类分析:一种通过将相似的数据划分到特定的组中以简化大规模数据集的方法。 14. 无监督分析法:这种方法没有内建的分类标准,组的数目和类型只决定于所使用的算法 和数据本身的分析方法。 15. 有监督分析法:这种方法引入某些形式的分类系统,从而将表达模式分配到一个或多个 预定义的类目中。 16. 微阵列芯片:将探针有规律地排列固定于载体上,与标记荧光分子的样品进行杂交,通 过扫描仪扫描对荧光信号的强度进行检测,从而迅速得出所要的信息。 17. 虚拟消化:是基于已知蛋白序列和切断酶的特异性的情况下进行的理论酶切(课件定 义)。是在已知蛋白质序列和蛋白外切酶之类切断试剂的已知特异性的基础上,由计算机进行的一种理论上的蛋白裂解反应。 18. 质谱(MS)是一种准确测定真空中离子的分子质量/电荷比(m/z)的方法,从而使分子质量 的准确确定成为可能。 19. 分子途径是指一组连续起作用以达到共同目标的蛋白质。 20. 虚拟细胞:一种建模手段,把细胞定义为许多结构,分子,反应和物质流的集合体。 21. 先导化合物:是指具有一定药理活性的、可通过结构改造来优化其药理特性而可能导致 药物发现的特殊化合物。就是利用计算机在含有大量化合物三维结构的数据库中,搜索能与生物大分子靶点匹配的化合物,或者搜索能与结合药效团相符的化合物,又称原型物,简称先导物,是通过各种途径或方法得到的具有生物活性的化学结构
生物信息学分析方法
核酸和蛋白质序列分析 蛋白质, 核酸, 序列 关键词:核酸序列蛋白质序列分析软 件 在获得一个基因序列后,需要对其进行生物信息学分析,从中尽量发掘信息,从而指导进一步的实验研究。通过染色体定位分析、内含子/外显子分析、ORF分析、表达谱分析等,能够阐明基因的基本信息。通过启动子预测、CpG岛分析和转录因子分析等,识别调控区的顺式作用元件,可以为基因的调控研究提供基础。通过蛋白质基本性质分析,疏水性分析,跨膜区预测,信号肽预测,亚细胞定位预测,抗原性位点预测,可以对基因编码蛋白的性质作出初步判断和预测。尤其通过疏水性分析和跨膜区预测可以预测基因是否为膜蛋白,这对确定实验研究方向有重要的参考意义。此外,通过相似性搜索、功能位点分析、结构分析、查询基因表达谱聚簇数据库、基因敲除数据库、基因组上下游邻居等,尽量挖掘网络数据库中的信息,可以对基因功能作出推论。上述技术路线可为其它类似分子的生物信息学分析提供借鉴。本路线图及推荐网址已建立超级链接,放在北京大学人类疾病基因研究中心网站(https://www.360docs.net/doc/8918082566.html,/science/bioinfomatics.htm),可以直接点击进入检索网站。 下面介绍其中一些基本分析。值得注意的是,在对序列进行分析时,首先应当明确序列的性质,是mRNA序列还是基因组序列?是计算机拼接得到还是经过PCR扩增测序得到?是原核生物还是真核生物?这些决定了分析方法的选择和分析结果的解释。 (一)核酸序列分析 1、双序列比对(pairwise alignment) 双序列比对是指比较两条序列的相似性和寻找相似碱基及氨基酸的对应位置,它是用计算机进行序列分析的强大工具,分为全局比对和局部比对两类,各以Needleman-Wunsch 算法和Smith-Waterman算法为代表。由于这些算法都是启发式(heuristic)的算法,因此并没有最优值。根据比对的需要,选用适当的比对工具,在比对时适当调整空格罚分(gap penalty)和空格延伸罚分(gap extension penalty),以获得更优的比对。 除了利用BLAST、FASTA等局部比对工具进行序列对数据库的搜索外,我们还推荐使用EMBOSS软件包中的Needle软件(http://bioinfo.pbi.nrc.ca:8090/EMBOSS/),和Pairwise BLAST (https://www.360docs.net/doc/8918082566.html,/BLAST/)。以上介绍的这些双序列比对工具的使用都比较简单,一般输入所比较的序列即可。 (1)BLAST和FASTA FASTA(https://www.360docs.net/doc/8918082566.html,/fasta33/)和BLAST (https://www.360docs.net/doc/8918082566.html,/BLAST/)是目前运用较为广泛的相似性搜索工具。这两
最新生物信息学学习心得
生物信息学学习心得 第一篇:生物信息学 生物信息学是上世纪90年代初人类基因组计划(hgp)依赖,随着基因组学、蛋白组学等新兴学科的建立,逐渐发展起来的生物学、数学和计算机信息科学的一门交叉应用学科。目前生物信息学的研究领域主要包括基于生物序列数据的整理和注释、生物信息挖掘工具开发及利用这些工具揭示生物学基础理论知识等领域。生物信息学作为新型交叉应用学科,可以依托本校已有的计算机科学、信息学、生物学和数学等学科优势,充分展现投入少、见效快、起点高的特色,推动学校学科建设和本科教学水平。 本实验指导书中的8个实验均设计为综合性开发实验,面向生物信息学院全体本科学生和研究生,以及全校对生物信息学感兴趣的其他专业学生开放。生物信息学实验室将提供系统的保障,包括采用mail服务器和linux帐号管理等进行实验过程管理和支持。限选《生物信息学及实验》的生物技术专业本科生至少选择其中5个实验,并不少于8个学时,即为课程要求的0.5个学分。其他选修者按照课时和学校相关规定计算创新学分。实验一熟悉生物信息学网站及其数据的生物学意义 实验目的:
培养学生利用互联网资源获取生物信息学研究前沿和相关数据的能力,熟悉生物信息学相关的一些重要国内外网站,及其核酸序列、蛋白质序列及代谢途径等功能相关数据库,学会下载生物相关的信息数据,了解不同的数据文件格式和其中重要的生物学意义。 实验原理: 利用互联网资源检索相关的国内外生物信息学相关网站,如:ncbi、sanger、tigr、kegg、sble、中科院北京基因组研究所、北大生物信息 学中心等,下载其中相关的数据,如fasta、genbank格式的核算和蛋白质序列、pathatdb格式化库文件,并输入blast命令进行计算,获得结果文件。 实验内容: 1. 向网上blast服务器提交序列,得到匹配结果; 2. 本地使用blast,格式化库文件,输入命令行得到匹配结果;