Abemaciclib合成路线图解
阿巴卡韦合成路线图解

[3] Vince R, Lackey JW. Synthesis of purine substituted cyclopentene derivatives[P]. WO: 9115490, 1991-10-17. (CA 1992, 116: 59910)
[4] Wallis CJ, Jones MF. Process for preparing a chiral nucleoside analogue[P]. WO: 9924431, 1999-05-20. (CA 1999, 130: 325348)
[6] Olivo HF, Yu J. Practical enantiodivergent syntheses of both enantiomers of carbovir, 1592U89 and six-membered ring analogues[J]. J Chem Soc Perkin Trans, 1998, 3: 391-392.
[2] Hodgsom DM, Witherington J, Moloney BA. Concise racemic and highly enantioselective approaches to key intermediates for the syntheses of carbocyclic nucleosides and pseudoribofuranoses:formal syntheses of carbovir[J]. J Chem Soc Perkin Trans I, 1994, 3373.
药物Abemaciclib(玻玛西林)合成检索总结报告
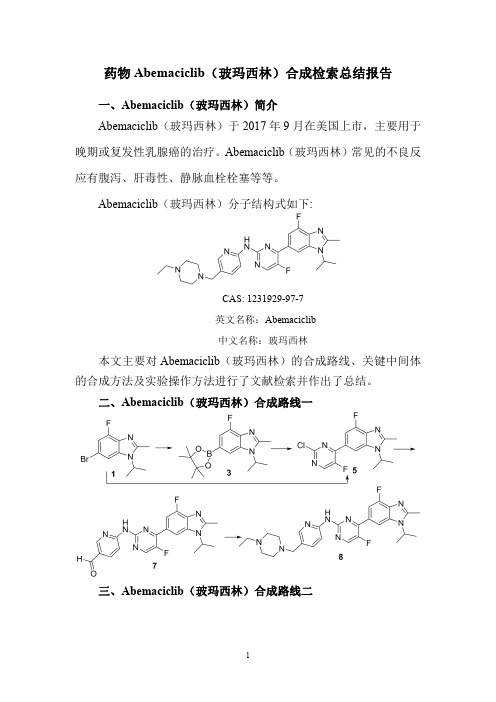
药物Abemaciclib(玻玛西林)合成检索总结报告一、Abemaciclib(玻玛西林)简介Abemaciclib(玻玛西林)于2017年9月在美国上市,主要用于晚期或复发性乳腺癌的治疗。
Abemaciclib(玻玛西林)常见的不良反应有腹泻、肝毒性、静脉血栓栓塞等等。
Abemaciclib(玻玛西林)分子结构式如下:CAS:1231929-97-7英文名称:Abemaciclib中文名称:玻玛西林本文主要对Abemaciclib(玻玛西林)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Abemaciclib(玻玛西林)合成路线一三、Abemaciclib(玻玛西林)合成路线二四、Abemaciclib (玻玛西林)合成路线一检索总结报告(一)Abemaciclib (玻玛西林)中间体3的合成(路线一)合成方法实验步骤参考文献操作方法一DMSO (100mL)was added to a 500mL one-necked flask and the compound of formula 1(20.0g,73.80mmoL),bis (pinacolato)diboron 2(27.6g,108.69mmol),tricyclohexylphosphine (3.53g,12.61mmol),Potassium acetate (21.3g,217.38mmol).Nitrogen gas was added to the reaction flask after the rapid addition of palladium acetate (1.5g),nitrogen PaulProtect the next open heating to 90o C.After 3hours the reaction flask was cooled to room temperature and the reaction mixture was poured into 700mL of water,mixed thoroughly and subtracted PressureCN107266421;(2017);(A)Chinesefiltration,the filter cake was washed twice with100mL of water and dried to give a light brown solid.Crude was added 50mL petroleum ether,10mL Ethyl acetate beating10min, vacuum filtration to give a white solid3(18.8g,yield80.3%)操作方法二Under nitrogen protection,palladium acetate(28mg)and tricyclohexylphosphine(54.3mg)was added into thesolution of6-bromo-4-fluoro-1-isopropyl-2-methyl-H-benzo[d]imidazole1(300mg,1.10mmol),bis(pinacolato)-diboron2(422mg,1.70mmol)and potassium acetate(326mg,3.3mmol)in anhydrous dimethyl sulfoxide(DMSO,5mL),and the reaction was carried out at100°C.undernitrogen protection for2hours.After cooling to roomtemperature,the reaction was filtered on Celite,the filtercake was washed with ethyl acetate,the filtrate was washedwith brine,dried over anhydrous sodium sulfate,the filtratewas concentrated under reduced pressure,and the filtrate wasseparated on column chromatography(eluant:petroleumether/ethyl acetate(v/v)=3:1),to affard260mg of a paleyellow solid3.US2019/152954;(2019);(A1)English操作方法三A suspension of6-bromo-4-fluoro-2-methyl-1-(propan-2-yl)-1H-benzimidazole(1)(90g,331.95mmol),bis(pinacolato)-diboron(2)(126g,498mmol),AcOK(80g,815.15mmol), tricyclohexylphosphine(14g,49.8mmol),and Pd(OAc)2(7.45g,33.2mmol)in DMSO(1.0L)was sparged with N2and then stirred at100°C.for16h.TLC analysis(1:1petroleum ether/EtOAc)showed complete consumption ofthe starting material.The black suspension was poured intoH2O(3.0L)and extracted with EtOAc(2*3L).Thecombined organic phases were washed with brine,dried overNa2SO4,filtered,and concentrated.The residue was purifiedby flash chromatography(Biotage,1.0kg,0-40%EtOAc/petroleum ether)to provide4-fluoro-2-methyl-1-(propan-2-yl)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzimidazole(3)(77g,73%yield)as a yellow solid.US2019/330196;(2019);(A1)English操作方法四The6-Bromo-4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole1(9.0g,33.2mmol),Bis-boronic acid pinacolester2(0.65g,49.8mmo1),palladium acetate(840mg), tricyclohexylphosphine(1.63g)and potassium acetate(9,78g,99.8mmo1)were added to60mL of DMSO and thenitrogen was allowed to warm to80°C for6h.The organicphase was extracted with EA,and the organic phase waswashed with EA.The organic phase was dried withanhydrous sodium sulfate and filtered through the filtrate.The filtrate was concentrated and purified by silica gelCN104910137;(2017);(B)Chinese;EP3091008;(2016);(A1)English。
甘油磷脂的生物合成(思维导图)
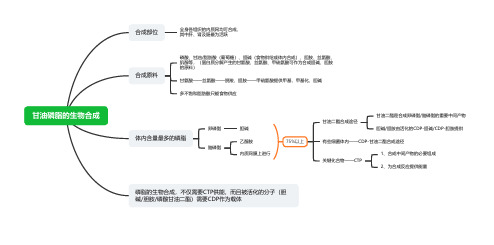
甘油磷脂的生物合成合成部位全身各组织的内质网均可合成,
其中肝、肾及肠最为活跃
合成原料
磷酸、甘油/脂肪酸(葡萄糖)、胆碱(食物供给或体内合成)、胆胺、丝氨酸、
肌醇等,(蛋白质分解产生的甘氨酸、丝氨酸、甲硫氨酸可作为合成胆碱、胆胺
的原料)
甘氨酸——丝氨酸——脱羧,胆胺——甲硫氨酸提供甲基,甲基化,胆碱
多不饱和脂肪酸只能食物供应
体内含量最多的磷脂
卵磷脂胆碱
脑磷脂
乙醇胺
内质网膜上进行
75%以上
甘油二酯合成途径
甘油二酯是合成卵磷脂/脑磷脂的重要中间产物
胆碱/胆胺由活化的CDP-胆碱/CDP-胆胺提供
有些细菌体内——CDP-甘油二酯合成途径
关键化合物——CTP
1、合成中间产物的必要组成
2、为合成反应提供能量
磷脂的生物合成,不仅需要CTP供能,而且被活化的分子(胆
碱/胆胺/磷酸甘油二酯)需要CDP作为载体。
泊马度胺合成路线

泊马度胺合成路线泊马度胺是一种重要的有机化合物,具有广泛的应用价值。
它可以用于制备药物、染料、涂料等多种化学品,因此其合成路线备受关注。
本文将介绍一种常用的泊马度胺合成路线。
泊马度胺的合成路线主要包括以下几个步骤:第一步:苯甲醛和丙二酸二乙酯反应,生成苯甲基丙二酸二乙酯。
第二步:苯甲基丙二酸二乙酯和氨水反应,生成苯甲基丙二酸二乙酰胺。
第三步:苯甲基丙二酸二乙酰胺和氯化亚砜反应,生成苯甲基丙二酸二乙酰亚胺。
第四步:苯甲基丙二酸二乙酰亚胺和氨水反应,生成泊马度胺。
这个合成路线的优点在于,反应条件温和,反应物易得,产率高,适用于大规模生产。
下面将对每个步骤进行详细介绍。
第一步:苯甲醛和丙二酸二乙酯反应,生成苯甲基丙二酸二乙酯。
这个步骤通常在酸性条件下进行,反应温度为室温至60℃。
反应物的摩尔比为1:1.2。
反应后,通过蒸馏纯化得到产物。
第二步:苯甲基丙二酸二乙酯和氨水反应,生成苯甲基丙二酸二乙酰胺。
这个步骤通常在碱性条件下进行,反应温度为室温至60℃。
反应物的摩尔比为1:1.5。
反应后,通过蒸馏纯化得到产物。
第三步:苯甲基丙二酸二乙酰胺和氯化亚砜反应,生成苯甲基丙二酸二乙酰亚胺。
这个步骤通常在室温下进行,反应物的摩尔比为1:1。
反应后,通过蒸馏纯化得到产物。
第四步:苯甲基丙二酸二乙酰亚胺和氨水反应,生成泊马度胺。
这个步骤通常在室温下进行,反应物的摩尔比为1:1。
反应后,通过蒸馏纯化得到产物。
总的来说,泊马度胺的合成路线简单、高效,适用于工业化生产。
但是,这个合成路线也存在一些问题,比如反应物的纯度要求较高,反应条件需要严格控制等。
因此,在实际应用中需要根据具体情况进行调整和改进。
2024届浙江省嘉兴市高三下学期二模化学试题及答案

2024年高三教学测试化学试题卷(2024.04)可能用到的相对原子质量:H-1 Li-7 C-12 N-14 O-16 Na-23 Si-28 S-32 Cl-35.5 K-39 Mn-55选择题部分一、选择题(本大题共16小题,每小题3分,共48分。
每小题列出的四个备选项中只有一个是符合题目要求的,不选、多选、错选均不得分)1.下列物质中含有共价键的盐是( )A .32CH CONHB .43NH NOC .2CaClD .NaOH2.工业上将2CO 通入到3NH 的饱和NaCl 溶液中制取3NaHCO ,下列说法不正确的是( )A .食品膨松剂的成份之一是3NaHCOB .-3HCO 电离出+H 使3NaHCO 溶液显酸性C .3NaHCO 不稳定,受热容易分解D .盐酸与3NaHCO 反应放出大量气泡并吸收热量3.下列表示不正确的是()A .2CO 的电子式:O C O∶∶∶∶B .2H O 的价层电子对互斥模型:C .Cl Cl —的p-p σ键的模型:D .的名称:1,3,4-三甲苯4.高铁酸钠(24Na FeO )是一种新型绿色消毒剂,主要用于饮用水处理,制备的一种方法其原理:-3+-2--423ClO +2Fe +10OH 2FeO +3Cl +5H O ,下列说法不正确的是()A .3+Fe 是还原剂B .2H O 既不是还原产物也不是氧化产物C .生成2-41mol FeO ,转移6mol 电子D .氧化产物与还原产物的物质的量之比为2:35.室温下,下列各组离子在指定溶液中一定能大量共存的是( )A .-10.1mol L H NaO ⋅溶液:++--2Na K ClO AlO 、、、B .()2+-1c Fe =1mol L ⋅的溶液:[]3-++-64K H Fe(CN)MnO 、、、C .水电离产生的()+-13-1c H =10mol L ⋅的溶液:+2+2--43Na Ca CrO HCO 、、、D .使甲基橙变红的溶液:2++-2-433Cu NH NO SO 、、、6.镁及其合金是用途很广的金属材料。
礼来乳腺癌试验药物Abemaciclib(Bemaciclib)的合成

Eli Lilly乳腺癌试验药物Abemaciclib (Bemaciclib)的合成背景介绍:近年来以抑制抑制细胞周期依赖性激酶4、6 (CDK4/6)为机理的乳腺癌药物受到了各方面的关注,也成为了各大医药巨头竞相开发的领域,其中包括以辉瑞,诺华和礼来三家公司为代表的新药palbociclib,Ribociclib (LEE011)和Abemaciclib(LY2835219).前面已经介绍了palbociclib,Ribociclib。
其中,palbociclib 是一种实验性、口服、靶向性制剂,能够选择性抑制细胞周期蛋白依赖性激酶4和6(CDK4/6),恢复细胞周期控制,阻断肿瘤细胞增殖。
此前,FDA于2013年4月授予palbociclib治疗晚期或转移性ER+/HER2-乳腺癌的突破性疗法认定。
争对手诺华(Novartis)已于去年11月将其CDK4/6抑制剂LEE011推进至III 期研究,礼来(Eli Lilly)的药物Abemaciclib(LY2835219)的推进则相对缓慢,研究进程落后于辉瑞和诺华。
据分析,至2020年,Palbociclib的销售额达29.50亿美元,而Abemaciclib的销售额则是6.51亿美元。
合成路线:酰胺化合物S-2在POCl3的作用下,形成氯亚胺中间体接受苯胺化合物S-1的进攻得到化合物S-3,S-3在碱性条件下,拔去质子,经过亲核芳香取代反应得到苯并咪唑化合物S-4,随后S-4芳基溴通过Pd催化和B2Pin2反应,制备得到芳基片那醇脂S-5,随后和氯代嘧啶化合物经过钯催化的Suzuki反应得到偶联产物S-7待用。
仲胺化合物S-8和醛S-9经过缩合,还原得到胺类化合物S-10,随后S-10在Pd 催化下,以联苯型CyJohnPhos(结构见后文)作为大位阻配体以及LiHMDS作为氮源制备2-氨基吡啶化合物S-11。
得到S-11以后,其和S-7通过Buchwald–Hartwig反应,得到化合物S-12,将化合物S-12做成甲磺酸盐及得到Abemaciclib (Bemaciclib)博客:/syntheticfuture 微博:/syntheticfuture参考路线:US20100160340。
脂肪代谢合成课件

乙酰CoA转运出线粒体过程图解
线粒体基质
内膜
HSCoA 柠檬酸合酶
柠檬酸
柠檬酸
H2O + 乙酰CoA
草酰乙酸
草酰乙酸
NADH + H+ 苹果酸脱氢酶
NAD+
苹果酸
苹果酸
丙酮酸羧化酶
ADP + Pi ATP + CO2
丙酮酸
丙酮酸
胞液
HSCoA + ATP 柠檬酸裂解酶
乙酰CoA+ADP+Pi NADH + H+ 苹果酸脱氢酶 NAD+
细胞中发生部位 酰基载体
二碳片段的加入与裂解方式
从头合成
细胞质 ACP-SH 丙二酰单酰CoA
β—氧化
线粒体 CoA-SH 乙酰CoA
电子供体或受体 酶系
原料转运方式 羟脂酰化合物的中间构型 对二氧化碳和柠檬酸的需求
NADPH
七种酶和一个蛋白质 组成复合物
三羧酸转运系统 D-型 需求
FAD、NAD+ 四种酶
肉碱穿梭系统 L-型
不需求
能量变化
消耗7个ATP和
产生106个ATP
反应途径中的4步反应比较
二、脂肪酸碳链的延长
★ 软脂酰CoA或软脂酸生成后,可在光滑内质 网及线粒体经脂肪酸碳链延长酶系的催化作用下, 形成更长碳链的饱和脂肪酸。
线粒体延长途径:基本上是β-氧化的
逆过程,只是NADPH 作为供氢体参与第 延 二次还原反应。 长
甘油磷脂(磷脂酰甘油):由甘 油构成的磷脂,是生物膜的主要 组分。
鞘氨醇磷脂:含鞘氨醇而不含 甘油的磷脂,是神经组织各种 膜(如神经髓鞘)的主要结构 脂之一。
阿布昔替尼化学结构-概述说明以及解释

阿布昔替尼化学结构-概述说明以及解释1.引言1.1 概述阿布昔替尼(Abemaciclib)是一种新型的抗癌药物,属于一类被称为“CDK4/6抑制剂”的药物。
CDK4和CDK6是细胞周期调节蛋白激酶,它们在细胞生长和增殖中起着重要的调控作用。
阿布昔替尼的化学结构与其他CDK4/6抑制剂有所不同,这使得它具有独特的药理特性和潜在的药效。
阿布昔替尼的化学结构中包含苯并三唑环和吡嗪环,并与CDK4/6结合,从而阻止其与细胞周期相关的底物结合,进而抑制肿瘤细胞的增殖。
与传统的化疗药物相比,阿布昔替尼能够更准确地靶向肿瘤细胞,并且对正常细胞的毒副作用较小。
这使得阿布昔替尼成为治疗某些癌症类型的有前景的药物。
阿布昔替尼的合成方法是通过有机合成化学反应来合成的。
具体来说,它是通过苯并三唑和吡嗪为原料,经过多步反应合成而成。
这种合成方法具有高效、高选择性和可控性的特点,能够在合成过程中控制反应的条件和操作,从而得到高纯度和高产率的阿布昔替尼。
目前,阿布昔替尼已经在临床试验中显示出良好的治疗效果,特别适用于乳腺癌、胰腺癌和肾透明细胞癌等恶性肿瘤的治疗。
它可以作为单药使用,也可与其他抗癌药物联合使用,以增强疗效。
在医药领域的前景中,阿布昔替尼被认为是一种具有重要且广泛应用的药物。
随着对其作用机制的深入研究和临床试验的不断推进,阿布昔替尼有望成为治疗多种癌症的首选药物之一。
未来,可以预见,阿布昔替尼的化学结构还有很大的发展潜力。
通过对其结构进行改造和修饰,可以进一步提高其抗肿瘤活性和药物代谢特性,使其更适合临床应用。
因此,对阿布昔替尼化学结构的研究和展望具有重要意义,并且值得进一步深入探索。
1.2文章结构文章结构本文分为引言、正文和结论三个部分。
引言部分主要是对阿布昔替尼的背景和重要性进行概述。
通过引入相关背景知识和对阿布昔替尼的简要介绍,让读者对接下来的内容有一个整体的认识。
正文部分是整篇文章的核心部分,主要包括阿布昔替尼的化学结构及特点、阿布昔替尼的合成方法和阿布昔替尼的应用领域三个方面的内容。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
F N
OB
N
O
24
DMF-DMA 87.9%
F 中间体15
OHC
F
NN
NN
N
8
H
27
N
74%
F N
H2N
CHO PdCl2, DPEPhos
99%
F
N
F
F
Cl N Cl F
N
7
N N
NN NN
N
PdCl2(PPh3)2 63.7% N N
Cl
N
Cs2CO3
Pd2(dba)3,Xantphos
N N
6
NO N Ot-Bu H
NH N
8
N Br
9
O H NaBH(OAc)3,DCM
98.2%
N N
10
LiHMDS, Pd2(dba)3, CyJohnPhos
N
49.1%
Br or Cu2O,NH3,54.5%
N N
N HNO3 NC NH2 NH2 86.2%
中间体7
N N
N NH HNO3
2) 以 1-乙基哌嗪(8)为起始原料,在三乙酰氧基硼氢化钠的存在下与 6-溴-3-吡啶甲醛(9)反应得到 10。 10 在以甲醇为溶剂、氧化亚铜存在的条件下,以液氨为氮源,得到中间体 7。或者以六甲基二硅基氨基 锂(LiHMDS)为氮源,在 2-(二环己基膦基)联苯(CyJohnPhos)、三(二亚苄基丙酮)二钯(Pd2(dba)3)的作用下, 10 转化成中间体 7 [7] [8] [9]。该法与 1)所示方法相比,明显的优点是步骤短,只需两步即可得到中间体 7,可大大缩短生产时间,利于工业化大生产。
Keywords
Abemaciclib, Synthesis, CDK4/6
Abemaciclib合成路线图解
张 浩1,陈奉泉2,张 剑1*
1潍坊医学院药学院医用化学教研室,山东 潍坊 2山东大学药学院药物化学教研室,山东 济南
收稿日期:2016年11月10日;录用日期:2016年11月25日;发布日期:2016年11月28日
*通讯作者。
文章引用: 张浩, 陈奉泉, 张剑. Abemaciclib 合成路线图解[J]. 药物化学, 2016, 4(4): 38-41. /10.12677/hjmce.2016.44005
张浩 等
摘要
Abemaciclib (Bemaciclib, LY-2835219)化学名称为N-[5-[(4-乙基-1-哌嗪)甲基]2-吡啶基]-5-氟-4-[4-氟 -1- 异 丙 基 -2- 甲 基 -1H- 苯 并 咪 唑 -6- 基 ]-2- 嘧 啶 胺 , 是 礼 来 公 司 研 发 的 细 胞 周 期 蛋 白 依 赖 性 激 酶 (cyclin-dependent kinase, CDK)4/6抑制剂。其单一用药对转移性乳腺癌有显著疗效,于2015年10月 被美国FDA授予“突破性疗法”认证。本文综述了Abemaciclib及关键中间体的合成。
40
张浩 等
3) 在碳酸铯、三(二亚苄基丙酮)二钯(Pd2(dba)3)、4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(Xantphos)共同 作用下,中间体 7 和中间体 25 发生 Buchwald–Hartwig 偶联反应得 Abemaciclib (1)。
2) 2,6-二氟苯胺(16)为起始原料,在氟乙腈/二氯甲烷的环境中,与三氟甲磺酸(TfOH)反应,得到化合 物 12。在三氯化磷、N,N-二异丙基乙胺(DIEA)存在的环境中,12 与 N-异丙基乙酰胺(21)发生脒化反应得 化合物 17。17 在氢化钠的碱性环境中自身环合,生成化合物 18。18 同 DMF-DMA 反应得中间体 15。
Open Access
Abstract
Abemaciclib (Bemaciclib, LY-2835219), whose chemical name is N-[5-[(4-ethyl-1-piperazinyl) methyl]-2-pyridinyl]-5-fluo-ro-4-[4-fluoro-2-methyl-1-(1-methylethyl)-1H-benzimidazol-6-yl]-2pyrimidinamine, is a significant inhibitor of the cyclin-dependent kinase (CDK) 4/6 researched by Lilly, showing single-agent activity in metastatic breast cancer. Abemaciclib was granted “Breakthrough therapy designation” by the FDA in October 2015. In this paper, we summarized the synthesis of Abemaciclib and key intermediate.
关键词
Abemaciclib,合成,CDK4/6
1. 引言
Abemaciclib (Bemaciclib, LY-2835219, 1)分子式:C27H32F2N8,CAS 登记号:1231929-97-7。该药作 为细胞周期蛋白依赖性激酶(CDK)4/6 抑制剂对转移性乳腺癌的治疗获得了 FDA“突破性疗法”认证[1] [2]。目前正处于转移性乳腺癌和非小细胞肺癌的Ⅲ期临床试验阶段[3]。Abemaciclib 抑制 CDK 4/cyclin D1 和 CDK 6/cyclin D1 的 IC50 值分别是 2.0 nM 和 9.9 nM [4],在转移性乳腺癌的临床试验中,与其他处于 临床试验阶段的 CDK4/6 抑制剂相比,其在限定剂量范围内的毒副作用是腹泻,而不是骨髓毒性[5]。本 文综述了 Abemaciclib (1)及关键中间体 7(5-[(4-乙基哌嗪-1-基)甲基]吡啶-2-胺)、15(6-(3-N,N-二甲基氨-2氟-2-丙烯酮-1-基)-4-氟-1-异丙基-2-甲基-1H-苯并咪唑)、25(6-(2-氯-5-氟-嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H苯并咪唑)的合成(图 1)。
12
F
16
21
PCl3, DIEA 84.6%
O
F
F
NN FH
17
O Cl
19
NH2
K2CO3 65.37%
OO O
NH2 91.35%
20
O N H
21
F NH2
Br
F
POCl3 Br 99%
FH NN F 22
t-BuOK 99% Br
NaH 83.3%
O
F
N
N F 18
F N N
23
B2pin2 PCy3, KOAc
87.3%
H N
Abemaciclib 1 F
中间体25
Figure 1. Graphical Synthetic Routes of Abemaciclib (1) 图 1. Abemaciclib (1)的合成路线图解
39
张浩 等
2. 中间体 7 的合成
1) 以 6-氨基烟酸乙酯(2)为起始原料,以乙腈为溶剂,经 4-二甲氨基吡啶(DMAP)催化 Boc 酸酐保护 氨基得 3。氢化铝锂将 3 还原为 4。在三乙胺(TEA)存在的条件下,4 与甲基磺酸酐反应生成化合物 5。在 碳酸铯存在的条件下,以 DMF 为溶剂,5 与 1-乙基哌嗪(8)反应得化合物 6。6 在二氧六环和盐酸的环境 中脱去 Boc 保护基,得到中间体 7 [6]。该法明显的缺点是步骤长、耗时长,并且在用氢化铝锂还原酯基 时,需要氮气保护,操作繁琐,不适合进行工业化大生产。
Hans Journal of Medicinal Chemistry 药物化学, 2016, 4(4), 38-41 Published Online November 2016 in Hans. /journal/hjmce /10.12677/hjmce.2016.44005
Received: Nov. 10th, 2016; accepted: Nov. 25th, 2016; published: Nov. 28th, 2016 Copyright © 2016 by authors and Hans Publishers Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). /licenses/by/4.0/
3. 中间体 15 的合成[10]
1) 4-氨基-3,5-二氟苯乙酮(11)为起始原料,与氟化剂 1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟 硼酸)盐(F-TEDA-BF4)反应生成化合物 12。12 中的活泼亚甲基同 N,N-二甲基甲酰胺二甲缩醛(DMF-DMA) 反应,得到 13。13 与 N-异丙基乙酰胺(21)在 TEA、三氯化磷存在时发生脒化反应,生成化合物 14。14 在叔丁醇钾的碱性环境中自身环合,生成中间体 15。
N NH2 (Boc)2O, DMAP
H N N Ot-Bu LiAlH4, THF
EtOOC
73.8% EtOOC
O
59.5% HO
2
3
O OO O
H
SS
N NHale Waihona Puke Ot-BuOO 4
TEA, DCM 91.9%
O S OO
H
N N Ot-Bu
8
O 5
Cs2CO3, DMF 90,4%
HCl 91.2%
N H
NH2