示差分光光度法测定水样中非那西汀的含量
【doc】盐酸氟西汀胶囊溶出度测定

盐酸氟西汀胶囊溶出度测定method0f0uaLLtvcontrolforcompoundbeazoicacidpowder.METHODS:Withoutseparat ion,thefivecontentsofthepowderweredetermineddirect[ybyapplyingunitedmethodsofpolarimetry,chemicalquantitativeangtlysisand1Lne arcombinationofabsorbances.RESULTS:The3ve[agerec0iesfbenzoicadd.furacilin,tannicacid,salicylicacidaadboricacidwere9999,100.07 ,99.96,i00.06aad10074re sDectLvelvTheRSDfortheahovementionedfivecomponents;vere0.5O.033.0.92,0.53an d0s0NrespectivelyCONCIU SIONThemethodformedcharacterizedbysimplicityaadremarkab[ereproducibilityThere sultsobtainedweresatisfiedandcouldbeusedinpractiea[analysis.KEYWORDScompoundbenzoicacidpower;polar[meto';LLnearcombinationmethodofa bsorbances;chemicalquantitationana]ys[s~con一^mJ,natio"∥/7一午盐酸氟西汀胶囊溶出度测定//,/上海中西蔷份洪上誓200065)Y2////(上海中西药业股份有限套司上海\/摘要目的:采用紫外分光光度法洲定国产盐酸氟西汀肢囊的溶出度.方法:腭中华人民共和国药典1995年版二部附录所速的溶出度洲定转蓝法,转速为75r/rain,水为溶剂.取样时间为30min,剥定波长为226nm._测定结果用空胶壳作空白校正结果:盐酸氟西汀水溶液在4.5~22.5f.Lg,/m]琅度范围内暧收度与琅度呈良好线性关系,回归方程Y一003576X一0O01.r一0.9999回收率为1O0.1,CV=0.46—5),瘩出限度太于80.结论:丰方法具有摊作简单,快连,鲒果准璃的优点,适用于盐酸氟西汀腔囊生产的质量控制关键词盐酸氟西汀肢囊溶出度紫外分光光度法盐酸氟西汀胶囊是一种新型抗抑郁药,我公司于1993年起研制开发作者采用紫外分光光度法测定国产盐酸氟西汀胶囊的溶出度.1仪器与药品1.1仪器RC一3B药物溶出仪(天津大学无线电厂生产);UV一201OPG型紫外分光光度计(日本岛津).1.2药品进口盐酸氟西汀胶囊(美国礼莱公司生产,批号:B0183CE);上海中西药业股份有限公司生产的盐酸氟西汀(批号:940702)及其胶囊(批号:941213,941214,941215).2方法与结果根据中华人民共和国药典1995年版二部附录中药品固体制剂溶出度的规定.我们耐用紫外分光光度法测定盐酸氟西汀胶囊溶出度进行的试验及其结果如下.2.1测定波长取盐酸氟西汀适量,用水和0.1mol/L盐酸渡分别制成每1ml占1O#g的溶液,用紫外分光光度计于200~400nm波长处扫描结果,两溶液在226rim和260nm波长处均有吸收峰.由于在260nm波长处吸收度小,灵敏度低,故选用226nm作为测定波长.2.2溶荆取盐酸氟西汀胶囊,以水和0.1mot/L盐酸液为溶剂,困胶囊容易上浮,给操作带来不便,故按中华人民共和国药典1995年版二部附录溶出度测定转篮法分别于3,6,10, 上海医药1999年第20卷第9期25,45min取样测定结果表明,盐酸氟西汀胶囊在水和0.1mo[/L盐酸渡中溶出率基本相同.故采用水作为测定溶剂.2.3标准曲线精密称取经105℃干燥至恒重的盐酸氟西汀适量.加水制成0224mg/m[的标准储备渡.分别吸取该溶液2.0, 4.0,6.0,8.0,10.0ml,各置100ml量瓶中.加水至刻度.摇匀,用紫外分光光度计在波长226nm波长处测定吸收度. 并对吸收度和浓度作线性回归.结果表明,盐酸氟西汀水溶液在4.5~22.4/~g/ml范围内吸收度与浓度呈良好线性关系,回归方程Y0.03576X一0.001,r一0.9999t一5j2.4盐酸氟西汀水溶液的稳定性将上述配制溶液室温放置2,4,8,24小时后分别测定其吸收度结果表明,该溶液在24小时内稳定.2.5回收率试验取盐酸氟西汀,分别配制成盐酸氟西汀胶囊处方量80~120浓度的水溶液,再取上述溶液,分别加入相当于1粒盐酸氟西汀胶囊处方量的辅料,振摇,离心分离,取上清液,在226nm波长处分别测定上述两种溶液的吸收度,计算各对应浓度的回收率及平均回收率.结果表明.其平均回收率为100.1,CV一0.46,说明紫外分光光度法可用于本品的定量测定.2.6转速取盐酸氟西汀胶囊,以水为溶剂,测定波长为226nm.转速分别为75r/rain和100r/rain,接中华』,民共和国药典391995年版二部附录溶出度测定转篮法操作,30min取样测定结果表明,两种转速的盐酸氟西汀胶囊的溶出量基本一致.故确定以?5r/m[n为测定溶出度时的转速.27空胶壳对溶出度测定值的影响取空胶壳6粒,按前述方法,以水为溶剂.转速为75r/m[n,测定波长为226nm,30m[n时取样测定其吸收度.结果表明.空胺壳在226nm处的吸收度平均值为0.062(SD 0.79).约占测定盐酸氟西汀胶囊时吸收度的15,故应在后者的溶出度测得结果中加以扣除.28溶出限度取盐酸氟西汀胶囊.按前述方法于溶出开始后5,10,15,20,30,45,60m[n时分别取样10m[(每次取样后补充溶剂10m1)滤过,精密量取续滤液5ml,置10m[量瓶中,加水稀释至刻度,摇匀.用紫外分光光度计在波长226nm处测定各样品的吸收度.并将测定结果用空胶壳作空白校正.另取进口盐酸氟西汀胶囊,同法操作.计算各时间间隔盐酸氟西汀胶囊的溶出百分率.结果表明.国产盐酸氟西汀胶囊与进口盐酸氟西汀胶囊的溶出情况基本相同.国产盐酸氟西汀胶囊10min时溶出70左右.20min时溶出达g0左右.故取样时间确定为30min.溶出限度定为太于8O.2.9样品-删定取盐酸氟西汀胶囊,按照中华人民共和国药典1995年版二部附录溶出度测定转篮法,以900m[水为溶剂,转速75r/min.30m[n时取样10ml,滤过,精密量取续滤液5m1. 置10m]量瓶中.加水稀释至刻度,摇匀.用紫外分光光度计0/,I在波长226nm处测定样品的吸收度,并将测定结果用空胺壳作空白校正.计算出每一粒胺囊的溶出量,溶出限度应为标示量的80以上.3批样品测定结果见附表附表三批样品溶出度测定结果批号溶出度(】平均值SD123456【)(03讨论本品可用紫外分光光度法测定溶出度.测定波长为226nm由于胶壳在此波长下的平均吸收度为0.062,约占盐酸氟西汀胺囊吸收度的15,故样品测定值应用空胺壳测定值加以校正.经与进口盐酸氟西汀胶囊的溶出度比较.两种胶囊的溶出行为基本一致,30min时的溶出量均在80以上:本文所述的溶出度测定方法具有操作茼便,快速.结果准确的优点,适用于盐酸氟西汀胺囊生产的质量控制萃方法已收载于中华人民共和国卫生都部标准(试行j.参考文献L中华人民共和国卫生部药典委员会.中华人民共和国药翅l995年版二部.北京:化学工业出版杜,1995:附录66.HPLC测定复方氨基比林注射液中氨基比林,安替比林,巴比妥含量Z…海200050,(上海延安制药厂上海)f,摘要目的:探讨用HPI,C法测定氨基比林注射液中氧基比林,安营比林,巴比妥寺量的方法.方法:在~Bondparkc.柱用于谊品种生产过程中的质量控制关键词高效液相色谱法复方氨基比球注射液氨基比林安营比林巴比妥————————,一———一,————~——一复方氨基比林注射液是由氨基比林,安替比林,巴比妥配伍制成的解热镇痛药.根据地方药品标准,测定本品中这3个组份的含量.前者采用衍生分光光度法,后两者采用酸碱中和滴定法.作者试用HPLC浩,同时测定该注射液中3 个组份的含量.1仪器与试药l_19-嚣高效液相色谱仪(VTaters510泵,UV486检测器,81o色谱工作台),DU-7紫外分光光度仪(美国贝克曼).l_2试药上海医药1999年第20卷第9期。
液质联用法测定保健品中西地那非含量的不确定度评定

第35卷第16期2019年*月Vol.35No.16Aug.2019甘肃科技Gansu Science and Technology液质联用法测定保健品中西地那非含量的不确定度评定王小乔,许晓辉,张虹艳,张文,赵波,闫君,朱天虹,李晨曦(兰州市食品药品检验所,甘肃兰州730050)摘要:评定了液相色谱-质谱联用法测定补肾壮阳药中掺入西地那非含量的不确定度。
通过对测定过程中的不确定度来源进行分析,建立测量模型,并考察影响不确定度的因素,计算出各个分量的标准不确定度,再由标准不确定度得到合成不确定,最后得到扩展不确定度,计算出了测定的不确定度结果。
关键词:液质联用;西地那非;不确定度中图分类号:R286.0不确定度是用来表征被测量值所处的量值范围,是评测定结果是否准确的量化指标叫验过程中,受所用的对照、玻璃仪器、天平、测量仪器、操作人员的技术水因素的影响,测量的结果只能是,同时,检测准验准检测准验须建立测量不确定度评定的程序文件巴因此,不确定度评定到检测验的,是检测验质量$,不确定度评定、品、中成药、食品检测等领域,保健品中非法掺入药含量测定的不确定度$,丫品中非法药果,行危害了人民群众的身体健康?3%6@,家也陆续发布了一些保健品中非法药的补充检验方法$肾壮阳药,应用液质联用法对补肾壮阳药中非法西地那非含量测定的不确定进行了定,并计算出了测定不确定度结果,期液质联用法测定品中非法掺入药含量的不确定度定考$1材料与方法1.1试验仪器与设备液相色谱-质谱联用仪(安捷伦6460);天平(梅特勒MS105DU)枸缘酸西地那非对照品(纯度99.0%;批号(1305-007A1;来源:TLC)1.2试验方法1.2.1检验依据《国家食品药品监督管理局2016年第196号公告》1.2.2供试样制备称取约0.5g样品研细后转移至50mL容量瓶中,加乙@40mL,超声处理15min,冷至室温,用乙@稀释至刻度,摇匀,精密量取上述溶液1.00mL置10mL容量瓶中,用乙@定容至刻度,摇匀,过0.22!m的滤膜,待测。
复方乙酰水杨酸片中非那西丁含量测定

实验十四亚硝酸钠滴定法测定复方乙酰水杨酸片中非那西丁的含量一实验目的1.掌握亚硝酸钠标准溶液配制及标定的原理及方法;2.了解重氮化滴定的特点,掌握重氮化终点的判断;3.熟悉复方制剂含量测定的特点;3.掌握复方乙酰水杨酸片中非那西丁含量测定的原理及方法。
二实验原理对氨基苯磺酸是具有芳伯胺基的化合物,在酸性条件下,可与NaNO2发生重氮化反应而定量地生成重氮盐。
滴定终点,过量的NaNO2与碘化钾反应,生成碘,使淀粉碘化钾试纸变蓝,指示标定终点。
复方乙酰水杨酸片处方(77年版)乙酰水杨酸220g非那西丁150g咖啡因35g制成1000片本品每片中含乙酰水杨酸(C9H8O4)应为0.209-0.231g,含非那西丁(C10H13O2N)应为0.143-0.158g,含咖啡因(C8H10O2N4·H2O)应为31.5-38.5mg。
复方乙酰水杨酸片为白色片。
本品每片中含乙酰水杨酸(C9H8O4)与非那西丁(C10H13NO2)均应为标示量的95.0~ 105.0%;含咖啡因(C8H10N4O2·H2O)应为标示量的90.0~110.0%。
解热镇痛药。
用于发热、头痛、神经痛、牙痛、月经痛、肌肉痛、关节痛等。
复方乙酰水杨酸片中含有乙酰水杨酸(简称A)、非那西丁(简称P)和咖啡因(简称C)三种主要成分。
各成分之间性质差异大。
乙酰水杨酸为芳酸类药物,具酸性,,可用酸量法测定;非那西丁为芳酰胺类药物,具酰胺基,呈中性,但具潜在芳伯氨基,可将其在酸性条件下水解后用重氮化法测定;咖啡因为黄嘌呤类生物碱,碱性极弱,不能采用一般生物碱的含量测定方法,但可将其与碘发生定量沉淀以后,剩余的碘用硫代硫酸钠滴定从而求得咖啡因的含量。
含量测定反应1 乙酰水杨酸的测定:将残渣溶于中性纯,直接用0.1M氢氧化钠液滴定游离羧酸。
++C O O HO C O C H 3N a O HC O O N aO C O C H 3H 2O2 非那西丁的测定:第一步,水解,得到芳香伯胺:++H 2ONHO C 2H 5CO CH 3NH 2O C 2H 5CH 3CO O HH 2S O 4第二步,重氮化定量反应:N H 2O C 2H 5N a N O 2H C lN+O C 2H 5N C l-N a C lH 2O22++++3 咖啡因的含量测定: 以[B]代表咖啡因的结构C H 3N C C NCC H 3C NN OHC H 3O H 2O定量反应原理:2[B]+ 4I 2 + 2KI + H 2SO 4 2[[B]·HI ·I4] + K 2SO 4 (定量过量)过量的碘用硫代硫酸钠滴定:I 2 + 2Na 2S 2O 3 2NaI + Na 2S 4O 6三 仪器及试剂1.仪器:分析天平、干燥箱、回流装置、水浴锅、烧杯、量筒、酸式滴定管、玻璃棒等。
3.2.S.4原料药的质量控制修改版

. - - 3.2.S原料药13.2.S.4 原料药的质量控制13.2.S.4.1质量标准13.2.S.4.2分析方法123.2.S4.3分析方法的验证283.2.S.4.4批检验报告1243.2.S.4.5质量标准制定依据1283.2.S原料药3.2.S.4 原料药的质量控制3.2.S.4.1 质量标准表3.2.S.4.1-1 原料药质量标准. 可修编-. 可修编-. 可修编-. 可修编-临床研究用药品质量标准草案他达拉非TadalafeiTadalafil3C22H19N3O4389.40本品为(6R,12aR)-2,3,6,7,12,12a-六氢-2-甲基-6-[3,4-(亚甲基二氧)苯基]吡嗪并[1',2':1,6]吡啶并[3,4-b]吲哚-1,4-二酮。
按干燥品计算,含C22H19N3O4应为98.0%~102.0%。
【性状】本品为白色至类白色粉末;无臭无味。
在N,N-二甲基甲酰胺、二甲亚砜中易溶,略溶于四氢呋喃和乙二醇单甲醚,微溶于甲醇和乙腈,极微溶解于乙醇和异丙醇,在正己烷、正庚烷和水中不溶。
比旋度取本品,精密称定,加二甲亚砜溶解并定容稀释制成每1mL中约含10mg的溶液,依法测定(附录ⅥE),比旋度为+78°~+84°。
【鉴别】(1)取本品,加0.1%三氟乙酸水溶液-乙腈(1:1)制成每1 ml中约含10 μg的溶液,照紫外-可见分光光度法(附录Ⅳ A)测定,在221 nm、284 nm和291nm的波长处有最大吸收。
(2)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
(3)本品的红外光吸收图谱应与对照品的图谱一致。
【检查】有关物质取本品,精密称定,用0.1%三氟乙酸水溶液:乙腈=1:1(v:v)溶解并定容稀释制成每1 ml中约含0.4mg的溶液,作为供试品溶液;取他达拉非对照品,精密称定,用0.1%三氟乙酸水溶液:乙腈=1:1(v:v)配制成每1 ml中约含0.4μg的溶液,作为对照溶液。
药品快速检测车“中药非法添加枸橼酸西地那非近红外模型”刍议

N 8男宝胶 囊 ( ol 长春 经ห้องสมุดไป่ตู้ 药业 有 限公
司 2 0 00 ) C r lt n= 4 7 % 0 7 9 1 o eao 2 .0 i
N 3参茸 片( o1 葵花 药业集 团( 春 ) 伊 有
限公 司 2 00 0 ) C re tn=0 0 % 0 83 1 o l i ao .0
对2 8批样 品 , 利用理化鉴别 发现 8批 呈 阳性 , 利用 薄层色谱 发现 6批呈阳性 ; 利用 中药非 法添加枸 橼酸 西地那 非近红 外
模 型 筛 查 全 部 呈 阴 性 , 有 样 品 都 通 过 了 初 筛 。结 果 见 表 1 所 。 4 讨 论 与 分 析
分析纯 ( 安徽 省巢湖市药 品检验 所提供 ) 近 红外光谱 比对模 ; 型: 中药非法添加枸 橼 酸西地 那非 近 红外模 型 ( 国药品 生 中 物制 品检定所提供 ) 。 J
—1 0碘化 钾 溶 液 4 1轻 轻 倾 倒 于 薄 层 板 板 面 上 , 0m ) 日光 下 检
年, 为了配合中成药专项监督 检查 , 快速 筛查 中成 药制剂 中是 否非法添加化学物质 , 国家食 品药 品监督管理局 中国药 品生物
制 品检 定 所 。 为药 品 快 速 检测 车提 供 了 中药 非法 添 加 枸 橼 酸西 地那 非 近 红 外 模 型 J 。本 文 利用 理 化 鉴 别 法 、 层 色谱 法 对 此 薄
公 司 ) 。
我们在巢湖市 区域 内抽取补 肾壮 阳类 中药共计 7 2批 , 其 中1 7批胶囊剂样 品适 于 中药非法 添加枸橼 酸西地 那非 近红 外模型筛查 ,0批 片剂 样 品、 1 1批 丸剂 ( 药 ) 处理 后也 使 假 经
片剂和注射剂中药物含量测定

(1)吸收系数法
(2)对照法
98:83.对乙酰氨基酚的含量测定方法为:取本品约40mg,精密称定,置250ml量瓶中,加0.4%氢氧化钠溶液50ml溶解后,加水至刻度,摇匀,精密量取5ml,置100ml量瓶中,加0.4%氢氧化钠溶液10m1,加水至刻度,摇匀,照分光光度法,在257nm的波长处测定吸收度,按C8H9NO2的吸收系数( )为715计算,即得。若样品称样量为m(g),测得的吸收度为A,则含量百分率的计算式为(A)
排除 改用氧化电位稍低的氧化剂 硫酸亚铁 高锰酸钾滴定法 硫酸亚铁片 硫酸铈滴定法 氧化剂 氧化电位
KMnO4 + 1.51 V Ce(SO4)2 + 1.44 V HNO3 + 0.94 V FeCl3 + 0.77 V I2 + 0.54 V
A. B. C. D. E.
例:利血平的含量测定方法为: 对照品溶液的制备 精密称取利血平对照品20mg,置100ml量瓶中,加氯仿4ml使溶解,用无水乙醇稀释至刻度,摇匀;精密量取5m1,置50ml量瓶中,加无水乙醇稀释至刻度,摇匀,即得。 供试品溶液的制备 精密称取本品0.0205g,照对照品溶液同法制备。
例:丙酸睾酮注射液 含量测定 USP(21)柱色谱——UV法 ChP(2000)甲醇提取——HPLC法 例:碘化油注射液 含量测定
△
例:黄体酮注射液含量测定 ChP(2000)甲醇提取——HPLC法
(3)助溶剂:助溶剂有苯甲酸钠、枸橼酸钠等。没有一般分析方法,根据药物本身性质考虑,如咖啡因注射液,在酸性下用碘量法测定时,苯甲酸在酸性下析出,不产生干扰。
建议选则方法: 主药量大,附加成份不干扰 容量法或重量法测定 主药可溶于有机溶剂 有机溶剂提取后测定 主药量少 光谱法或色谱法
综合性实验报告(从茶叶中提取咖啡因)

广州大学化学化工学院本科学生综合性实验报告实验课程有机化学基础实验合成实验项目从茶叶中提取咖啡因专业班级学号姓名指导教师及职称开课学期2011 至2012 学年第二学期实验时间2012年3月27日APC主要成分简介阿司匹林阿司匹林的分子结构式普通命名法:乙酰水杨酸,邻乙酰水杨酸系统命名法:2-(乙酰氧基)苯甲酸分子式:C9H8O4相对分子质量:180.1阿司匹林是一种历史悠久的解热镇痛药,诞生于1899年3月6日。
用于治感冒、发热、头痛、牙痛、关节痛、风湿病,还能抑制血小板聚集,用于预防和治疗缺血性心脏病、心绞痛、心肺梗塞、脑血栓形成,应用于血管形成术及旁路移植术也有效。
性质描述:白色针状或板状结晶或粉末。
熔点135~140℃。
无气味,微带酸味。
在干燥空气中稳定,在潮湿空气中缓缓水解成水杨酸和乙酸。
在乙醇中易溶,在乙醚和氯仿溶解,微溶于水,在氢氧化钠溶液或碳酸钠溶液中能溶解,但同时分解。
该品1g能溶于300ml水5ml醇10-15ml醚或17ml氯仿。
生产方法及其他:水杨酸乙酰化而得:在反应罐中加乙酐(加料量为水杨酸总量的0.7889倍),再加入三分之二量的水杨酸,搅拌升温,在81-82℃反应40-60min。
降温至81-82℃保温反应2h。
检查游离水杨酸合格后,降温至13℃,析出结晶,甩滤,水洗甩干,于65-70℃气流干燥,得乙酰水杨酸。
非那西汀普通命名法:非那西汀或对乙氧基-N-乙酰苯胺系统命名法:4-乙氧基乙酰苯胺分子式:C10H13NO2相对分子质量:179.22非那西汀的分子结构式性质描述:又称非那西汀或对乙氧基-N-乙酰苯胺。
白色有光泽的鳞片状晶体或白色结晶粉末。
无臭。
味微苦。
在空气中稳定。
熔点135℃。
溶于乙醇、丙酮、乙酸、吡啶、氯仿等。
微溶于乙醚、苯和热水。
难溶于水。
饱和水溶液是中性反应。
由对乙氧基苯胺与醋酐、醋酸经乙酰化反应制得。
也可由对硝基氯苯经置换、还原和乙酰化制得。
是良好的解热镇痛药,常用于治疗头痛、发热、神经痛等。
差示扫描量热仪(DSC)在药物分析中的应用
差示扫描量热仪(DSC)在药物分析中的应用珀金埃尔默仪器(上海)有限公司朱兵刘继涛前言:药品的研发与生产必须监控其物化性质,如纯度、晶型、稳定性和安全性,以确保药物具有预期的药性。
众所周知,有机化合物包括药品常常具有多种结构及晶态,这势必影响到药品的加工条件、期稳定性、衰变及生物投递能力。
药品的最终组成中包含了多种活性组份以及它们之间相互作用而生成的产物,当然还有赋形剂、水分、药片涂层等,十分复杂。
因此对其全面的表征也变得越来越重要,其中最理想的测试方法之一就是热分析。
热分析具有用量少、方法灵敏、快速的特点,在较短的时间内可获得需要复杂技术或长期研究才能得到的各种信息。
差示扫描量热仪(DSC)是目前在医药领域应用最广的热分析仪之一,DSC通过测量药物热焓和温度随程序温控的变化,具体可以研究的信息如:z药物纯度z药物的多晶及亚稳态、无定形态的研究z优化冷冻干燥z脂质检测、蛋白质变性1. 药物纯度DSC在药物分析中最主要的应用之一是评估药物纯度。
自从上世纪六十年代商业DSC 产品出现以来,因DSC测定药物纯度快速、准确易于操作,这项技术已被广泛接受。
DSC 池体的响应时间和温度测量对于纯度的准确分析至关重要。
功率补偿型DSC因其炉体小(<1g),响应时间极快,而且其使用铂电阻测温精度高、准确好,因而非常适合纯度的准确测量。
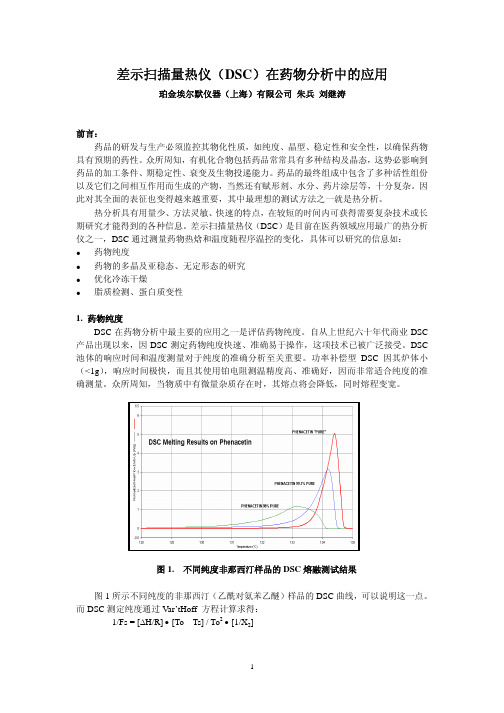
众所周知,当物质中有微量杂质存在时,其熔点将会降低,同时熔程变宽。
图1. 不同纯度非那西汀样品的DSC熔融测试结果图1所示不同纯度的非那西汀(乙酰对氨苯乙醚)样品的DSC曲线,可以说明这一点。
而DSC测定纯度通过Var’tHoff 方程计算求得:1/Fs = [∆H/R] • [To – Ts] / To2• [1/X2]式中Ts为样品的瞬间熔解温度,To为纯物质的熔点(°K);△H为纯物质的熔融热(J/g),X2为杂质样品中的摩尔分数;R为气体常数(8.314J/mole),Fs则为温度Ts时样品已熔化的分数,Fs=As/At,As为温度为Ts时已熔融部分的熔融热,At为总熔融热。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附:uv-2450使用说明
1、打开电源开关,打开电脑及软件,进入自检界面,进入自检画面。 2、单击确定开始自检,自检过程中不得打开样品室盖 3、完成自检后,单击波长扫描按钮,选择“方式”,设定波长及测量方式 (Abs) 4、将参比溶液放入参比池中,另一参比液放入样品池第一池内。点击自动 清零键清零。 5、取出样品池中参比,放入测量液,进行全波长扫描,找出最大吸收波长。 6、取出测量液,将标准曲线的五个试样放入测量池中,打开定量分析界面, 设定波长和测量方式Abs,测定各点吸收值,绘图 7、放入待测样品,测定
示差分光光度法测定水样 中非那西汀的含量
指导教师 姜桂兰
实验目的
1、通过标准曲线的绘制及样品溶液的测定,
了解示差分光光度法的原理及操作步骤 2、掌握UV-2450型紫外-可见分光光度计的使 用方法
实验原理
一、UV-2450型紫外-可见分光光度计基本组
成及其工作原理 二、示差分光光度法的原理
UV-2450紫外-可见分光光度计, 石英比色皿(1cm), 容量瓶(50ml), 移液管(1ml、10ml), 未知样品液。
实验方法及步骤
1.标准溶液的配制 非那西汀标准溶液(100μg/ml):准确称取非那西汀 0.0500g于小烧杯中,加少许乙醇溶解,转移到500ml容量 瓶中,用蒸馏水稀释至刻度,摇匀备用 2.吸收曲线绘制 移取5.00ml非那西汀标准溶液于50ml容量瓶中,蒸馏水 稀释至刻度,摇匀。以蒸馏水为空白,进行全波长扫描,绘 制吸收曲线,找出最大波长。
3.绘制工作曲线 取非那西汀标准溶液5ml,10ml,15ml,20ml,30ml分别 于5个50ml容量瓶中,用蒸馏水稀释至刻度,于最大波长处 测定吸光度值,以A对c作图绘制工作曲线。 4.样品测定 取10ml样品(浓度在40-60μg/ml)于50ml容量瓶中,蒸馏 水稀释至刻度,摇匀,于最大波长处测定吸光度值。
2、示差分光光度法的测定步骤: 1)采用浓度为Cs的标准溶液为参比溶液;
2)测定一系列Δ C已知的标准溶液的相对吸光度(Ar);
3)绘制Ar-Δ C工作曲线; 4)由测得试样溶液的相对吸光度Ar,x,即可从Ar-Δ C工作 曲线上求出Δ C; 5)根据下式求出试样浓度Cx : Cx=Cs+Δ C
试剂与器材
操作规程
1.打开电源开关,打开电脑及软件,进入自检界面,进入自检画面。 2.单击确定开始自检,自检过程中不得打开样品室盖 3.完成自检后,单击波长扫描按钮,选择“方式”,设定波长及测量方式 (Abs) 4.将参比溶液放入参比池中,另一参比液放入样品池第一池内。点击自动 清零键清零。 5.取出样品池中参比,放入测量液,进行全波长扫描,找出最大吸收波长。 6.取出测量液,将标准曲线的五个试样放入测量池中,打开定量分析界面, 设定波长和测量方式Abs,测定各点吸收值,绘图 7.放入待测样品,测定
例:若普通的分光光度法测得试样的T=5.0%, 配制一浓度 稍低的标准溶液S,测得T=10.0%, 二者之差为5%;用差示
法时,以此标准溶液S来调节仪器令T=100%,再来测定
试样X,可得T=50.0%, 二者之差为50%。这样,差示法相 当于把标尺扩大了10倍,测量读数的相对误差也就缩小 了10倍。
一、UV-2450型紫外-可见分光光度计基 本组成及其工作原理
1.分光光度法
利用紫外-可见吸收光谱对物质进行定性和定量分析的方 法就是紫外-可见分光光度法,属于分子吸收光谱,由分子 的外层电子跃迁产生,是宽带吸收光谱。 物质对光吸收遵循朗伯-比尔定律,即当入射光波长一 定时,待测溶液的吸光度A与其浓度和液层厚度成正比。
二、示差分光光度法原理及测量方法
1、示差分光光度法原理
示差分光光度法是应用于高含量组分测定的一种方法。
普通分光度法一般只适合于测定痕量组分,当待测组分 含量较高时,往往于朗伯-比尔定律产生偏离或因测得的吸光 度值超出适宜的的读数范围而引入较大的误差,使准确度降 低。而示差法却能很好的解决这一问题。两者的主要区别在 于参比溶液不同,普通的分光光度法采用不含已显色被测组 分的空白作参比溶液,而示差分光光度法则采用一合适浓度 的标准溶液作参比溶液。
3.UV-2450型紫外-可见分光光度计
日本岛津 单单色器、双光束方式、光电倍增管检测器 特点
1.低杂散光,高光通量,可对高浓度的样品不进行稀释直
接测定 2.应用广泛:可用于分析有机、无机化合物;DNA、酶 等生化样品;光学材料的特性 3.配备了功能强大的UVProbe软件,包含光谱测定、光 度测定、动力学测定和报告处理四大模块,从基本的测 定到研究解析都可通过它实现。
差示测定试液浓度(Cx)时,采用浓度稍低于试样的标准溶液 (Cs)作参比溶液调节仪器透光度读数为100%(A=0),再测定试样 溶液的吸光度(Ar称为相对吸光度),相对应的透光度(Tr)称为相 对透光度。 普通光度法以纯溶剂或空白试剂作参比溶液,测得标准溶液 及试液的吸光度分别为As和Ax,对应的透光度为Ts和Tx,根据比 尔定律 Ax=ε bCx,As=ε bCs Ar=Ax-As=ε b(Cx-Cs)=ε bΔ C 上式意义:在符合比尔定律测定浓度范围内,示差法测得的 相对吸光度(Ar)与被测溶液和参比溶液的浓度差(Cx-Cs,即Δ C) 或成正比,即可用于定量测定。此时试液的Tr,x=50%,令读数 落在适宜的范围内,提高了测定的准确度。
3)样品室
样品室放置各种类型的吸收池 (比色皿)和相应的池架附件。吸
收池主要有石英池和玻璃池两种。
在紫外区须采用石英池,可见区一 般用玻璃池。
4)检测器
利用光电效应将透过吸收池 的光信号变成可测的电信号,常 用的有光电池、光电管或光电倍 增管。
5)结果显示记录系统
检流计、数字显示、微机进行 仪器自动控制和结果处理
2.分光度计的基本组成
光源
单色器
样品室
检测器
显示
1)光源
在整个紫外光区或可见光谱区可以发射连续光谱,具
有足够的辐射强度、较好的稳定性、较长的使用寿命。
可见光区:钨灯作为光源,其辐射波长范围在320~ 2500 nm。
紫外区:氢、氘灯。发射185~400 nm的连续光谱。
2)单色器
将光源发射的复合光分解成单色光并可从中选出任一波 长单色光的光学系统。
实验结果
1.绘制非那西汀的吸收曲线
2.绘制工作曲线 3.可由显示器直接读出测定结果,记录
实验注意事项
1.仪器使用时要符合仪器工作环境的要求: 稳固的工作台 ,适宜的温度、湿度, 避免:日光直射、震动、强电场、强磁场。 2.仪器保养和维护方法 : 用软布稍微蘸取水,轻柔擦拭外表面,避免蘸取过多水而流 入仪器内部。 清除样品室内残留液体样品,防止蒸发,避免腐蚀样品室。 3.每半年进行一次波长准确度检查。 4.比色池的使用规则 1)手拿部位:毛玻璃面 2)用镜头纸擦拭透光面 3)用后及时清洗,放入比色池盒内。 5. 注意人身安全和仪器安全