常染色体隐性遗传病
医学遗传学名词解释及解答题

医学遗传学名词解释及解答题名词解释健康:是受人体遗传结构控制的代谢方式与人体的周围环境保持平衡。
遗传性疾病:因遗传因素而罹患的疾病。
先天性疾病:婴儿出生时即显示症状的疾病。
家族性疾病:一个家族有多个成员患同一种疾病。
核型:把人体某个细胞中的全部染色体按其大小、形态特征顺序排列所构成的图像称为核型。
嵌合体:同时存在两种或两种以上核型的细胞系的个体。
如:45,X/46,XX。
臂内倒位:染色体发生两处断裂后,中间的断片倒转180度后又重新连接;倒位部分不包括着丝粒而仅限于一臂之内。
臂间倒位:染色体发生两处断裂后,中间的断片倒转180度后又重新连接;倒位部分包括着丝粒脆性位点:染色体上特定位置出现的一种断裂点,但并非完全断裂,而是一种裂隙现象,即在此断裂点上可见有一细丝相连,或者有不着色的染色质相连接。
先证者:指在该家系中首先被确认的遗传病患者。
外显率:是指一群具有某种致病基因的人中,出现相应病理表现型的人数百分率。
外显率是一个全或无的概率,是个质的问题。
表现度:是指基因表达的程度,大致相当于临床严重程度。
分为重型,中型,轻型及顿挫型等。
表现度是个量的问题。
顿挫型:当一种畸形疾病或综合征的表现极为轻微而无临床意义时。
亲代印迹:是指同一基因会随着它来自父源或母源而有不同的表现。
亲缘系数:是指有共同祖先的两个人,在某一位点上具有同一基因的概率。
限性遗传:常染色体致病基因的表达仅限于一种性别受累者。
偏性遗传:虽非X连锁遗传的疾病,但在两性中的表达,其程度和频率均有不同。
显示杂合子:X伴性隐性遗传的女性杂合子表现出临床症状。
遗传异质性:有些临床症状相似的疾病,可有不同的遗传基础。
遗传早现:有些遗传病在世代传递过程中有发病年龄逐代超前和病情症状逐代加剧的现象。
拟表型:环境因素引起的疾病模拟了由遗传决定的表现型。
遗传度:遗传因素即致病基因在决定多基因遗传病表现型中所起作用的大小。
易感性:由遗传素质决定一个个体得多基因遗传病的风险。
人类遗传病的主要类型-编号[4873]
![人类遗传病的主要类型-编号[4873]](https://img.taocdn.com/s3/m/27bea7eb6294dd88d0d26b68.png)
常染色体隐性遗传病
苯丙氨酸
氨基转换
酪氨酸(正常)
缺少基因P,正常 积累 酶无法合成 苯丙酮酸(异常) 苯丙酮尿症
西红柿女孩
有一女孩患苯丙酮尿症,是真正意义上的不食 “人间烟火”,能吃的食物仅有西红柿。目前,低 苯丙氨酸饮食疗法是全世界治疗苯丙酮尿症唯一的 方法。所以,让女孩延续生命的食物全是化学产品, 近些的在北京,远些的就在日本和美国等地。
先天性疾病、家族性疾病与遗传病的关系
先天性疾病 家族性疾病
不一定是遗传病
遗传病
大多数遗传病是 先天性疾病
关 系
不一定是遗传病
例 子
遗传病:先天性愚 遗传病:显性 型,多指,白化病, 遗传病 苯丙酮尿症 非遗传病:由 非遗传病:胎儿 于饮食中缺乏 在子宫内受天花 维生素A,一家 病毒感染,出生 族中多个成员 时留有癜痕 患夜盲症
8、人类的遗传病中,当父亲是某病患者时,无论母亲 是否有病,他们子女中的女孩全部患此病,这种遗传 病最可能是( ) A.常染色体显性遗传病 B.常染色体隐性遗传病
C
C.X染色体显性遗传病
D.X染色体显隐性遗传病
9、在下列生殖细胞中,哪两种生殖细胞的结合会产 生先天愚型的男性患儿(A表示常染色体)? ①23A+X ②22A+X ③21A+Y ④22A+Y A.①和③ B.②和③
苯丙酮尿症
(二)多基因遗传病
概念: 由多对等位基因控制。常表现出家族性聚集现象, 且比较容易受环境影响。
目前已发现的多基因遗传病有100多种,如 唇裂 无脑儿 原发性高血压
青少年型糖尿病。
唇裂 多基因遗传病,
无脑儿
常染色体异常遗传病 猫叫综合征
原因:第5号 染色体部分 缺失。 症状:两眼距 离较远,耳位 低下,生长发 育缓慢,,存 在着严重的智 力障碍。患儿 哭声轻,音调 高,很像猫叫。
常染色体隐性遗传性耳聋

常染色体隐性遗传性耳聋王秋菊;袁永一【期刊名称】《听力学及言语疾病杂志》【年(卷),期】2016(024)004【总页数】4页(P421-424)【作者】王秋菊;袁永一【作者单位】解放军总医院耳鼻咽喉头颈外科,解放军耳鼻咽喉研究所北京100853;解放军总医院耳鼻咽喉头颈外科,解放军耳鼻咽喉研究所北京100853【正文语种】中文【中图分类】R764.44常染色体隐性遗传性耳聋是指与耳聋表型相关的基因位于常染色体上,此类耳聋只有在两个分别来自父母的等位基因均携带致病突变时才出现耳聋。
有关常染色体隐性遗传性耳聋的描述最早出现在16世纪,Schenck描述了一个家系中的多名兄弟姐妹均患有先天性重度耳聋,而他们的父母听力完全正常。
1853年,Wilde提出在常染色体隐性遗传性耳聋中前瞻性调查父母血亲关系十分重要。
但在1875年George Darwin否定了这一观点。
1877年,一项匿名研究指出,在Marsha's Vineyard,所有后代均来自共同的祖先,其中60%患有耳聋。
1880年Hartmann提供了耳聋显性及隐性遗传的证据,他也强调在隐性遗传性耳聋中研究父母近亲婚配的重要性。
常染色体隐性遗传性耳聋具有以下特征:①致聋基因位于常染色体,因而致聋基因的遗传与性别无关,即男、女性的患病机会均等;②系谱中通常看不到连续传递现象,往往是散发病例,但同胞中可有多人患病;③患者的双亲一般不患病,但都是致病基因的携带者;④患者的同胞有1/4的风险患病,患者表型正常的同胞中有2/3的概率为携带者;⑤患者的后代一般不发病,但一定是携带者;⑥近亲婚配时子女的发病风险显著提高,因为共同的祖先可能传递给他们共同的突变基因。
一般所指的常染色体隐性遗传性耳聋多指非综合征型耳聋(autosomal recessive nonsyndromic hearing loss,ARNSHL),耳聋是其唯一临床表现,大多数患者症状早发且程度较重。
高考生物必备知识点:遗传病的分类及特点

高考生物必备知识点:遗传病的分类及特点 近年来,随着医疗技术的发展和医药卫生条件的改善,人类的传染性疾病已经逐渐得到控制,而人类的遗传性疾病的发病率和死亡率却有逐年增高的趋势,已成为威胁人类健康的一个重要因素。
遗传病是因遗传物质的改变而引起的一类疾病,具有自身的发病特点,只有掌握其分类及遗传特点,才能有效防止或降低发病率,真正提高人类的健康水平。
1.分类 根据遗传物质存在部分不同可分为:细胞核遗传病、细胞质遗传病。
根据引起遗传病的原因又可将细胞核遗传病分为:单基因遗传病、多基因遗传病和染色体异常遗传病,对于单基因遗传病而言,又因基因存在的染色体及其显隐性分为:常染色体显性遗传病、常染色体隐性遗传病、X染色体显性遗传病、X染色体隐性遗传病和Y染色体遗传病;染色体异常遗传病又分为染色体结构异常疾病和染色体数目异常疾病,还可分为常染色体病和性染色体病。
2.遗传特点及常见病例 (1)单基因病①常染色体显性遗传病:遗传特点为连续遗传、无性别差异、家族性聚集等牲,如软骨发育不全、并指、多指、家庭性结肠息肉症等。
②常染色体隐性遗传病:遗传特点为隔代表现、无性别差异,如白化病、苯丙酮尿症、先天性聋哑、镰刀型细胞贫血病、婴儿黑蒙性白痴等③X染色体显性遗传病:遗传特点为连续遗传、交叉遗传、女性多于男性、男性患者的女儿均为患者,如抗维生素D佝偻病、遗传性肾炎等。
④X染色体隐性遗传病:遗传特点为隔代遗传、交叉遗传和男性多于女性,如血友病、进行性肌营养不良(假肥大症)、色盲症等。
⑤Y染色体遗传病:表现为限雄遗传、连续遗传的特点,如外耳道多毛症。
(2)多基因病由多对基因控制,呈家族聚集趋势,难以预测,无很好的预防方案,如唇裂、无脑儿、原发性高血压、青少年型糖尿病等。
(3)染色体病染色体数目异常疾病:如“21三体”综合征,又称为先天愚型、唐氏综合征,是由于21号染色体数目多了一条而形成的;性腺发育不良,又称为特纳氏综合征,是女性X染色体少一条导致的,形成原因是减数分裂异常形成了不含性染色体的雌雄配子与一个含X染色体的正常的异型配子结合形成的受精卵发育而成;克氏综合征:男性XXY ,形成此种个体的受精卵可能是由一个含XX染色体的雌配子与一个含Y染色体的雄配子而成,也可能是由一个正常的雌配子与另一个含XY染色体的雄配子结合而成。
ADA缺乏症的基因治疗

ADA缺乏症的基因治疗ADA缺乏症是SCID的主要病种,约占25%。
ADA缺乏症是人类基因治疗研究得较为成熟的一种常染色体隐性遗传性疾病。
由于ADA基因的克隆化早已完成,对其表达的调控认识得也比较清楚,ADA编码基因属于持家基因的范畴,因此人类历史上第一种以基因治疗的疾病就是:ADA缺乏症。
一、ADA缺乏症发病机制ADA有数种同功酶,在电泳上有二个带,分子量各带相异。
ADA广泛存在于各种组织与细胞中,在细胞中有三种同功酶,在组织中有四种同功酶。
除红细胞外,ADA在脾脏、皋丸、甲状腺、肺脏、肾脏次之;在心脏、肝脏、胃粘膜和肌肉中也存在。
其中以脾脏中的酶活性最高。
ADA在核酸代谢中具有重要作用。
它可催化腺苷变为次黄嘌呤核苷,后者再在嘌呤核苷磷酸化酶的作用下变为次黄嘌呤,又在黄嘌呤氧化酶作用下经黄嘌呤变为核酸代谢的最终产物一尿酸。
脾脏是重要的免疫器官,在人体免疫系统的发育中占有极为重要的地位。
当ADA缺乏时,细胞内腺苷及嘌呤核苷酸的水平增高,使乳清酸向乳清酸苷的转变被封闭,以致影响B淋巴细胞及免疫系统的发育,引起严重的体液、细胞免疫联合缺陷。
除此之外,ADA缺乏引起免疫缺陷的另一种可能的机制是存在一种抑制免疫的血清因子。
ADA基因异常时常可累及多种细胞,如红细胞等,但T、B淋巴细胞对腺苷及嘌呤核苷酸的敏感性远较其它种类的细胞敏感,因此这些核苷酸代谢产物的累积,对淋巴细胞的毒性和危害最大,因此导致T、B淋巴细胞的成熟过程及功能障碍,造成SCID的免疫病理状态。
二、ADA基因异常1983年人ADA的cDNA克隆化三家同时成功。
编码区有1089个核昔酸组成,编码的ADA 由363个氨基酸残基组成。
ADA基因组DNA的全长为3204个碱基对,由23个内含子和12个外显子序列组成。
ADA缺乏症是一种常染色体隐性遗传疾病,其基因的异常又分为纯合子型和杂合子型两种。
根据对多地区、多个ADA缺乏症ADA基因的序列的测定以及与正常人ADA序列的比较结果,目前认为ADA基因序列点突变至少发生在8个位点上。
苯丙酮尿症诊疗指南【2019版】

90. 苯丙酮尿症概述苯丙酮尿症(phenylketonuria,PKU)是由于苯丙氨酸羟化酶(phenyalanine hydroxylase,PAH)缺乏引起血苯丙氨酸(phenylalanine,Phe)浓度增高,并引起一系列临床症状的常染色体隐性遗传病。
苯丙酮尿症是高苯丙氨酸血症的主要类型。
病因和流行病学PAH 基因变异导致PAH 活性降低或缺乏是PKU 的主要病因。
苯丙氨酸是人体必需氨基酸,其代谢所需的苯丙氨酸羟化酶(PAH)活性降低或缺乏,使苯丙氨酸不能转化为酪氨酸(tyrosine,Tyr),酪氨酸及其他正常代谢产物合成减少,血液中Phe 含量增加,影响中枢神经系统发育。
同时次要代谢途径增强,生成苯丙酮酸、苯乙酸和苯乳酸,并从尿中大量排出,苯乳酸使患儿的尿液具有特殊的鼠尿臭味。
PAH 缺乏症发病率在不同种族和地区有差异。
爱尔兰约为1/4500,北欧、东亚约为1/10 000,日本约为1/143 000。
我国平均发病率为1/11 800。
根据血Phe 浓度将PAH 缺乏症分为:轻度HPA(120~360μmol/L) 、轻度PKU(360~1200μmol/L)、经典型PKU(≥1200μmol/L)。
临床表现PKU 患儿在新生儿期多无临床症状,出生3~4 个月后逐渐出现典型症状,1 岁时症状明显。
出生数月后因黑色素合成不足,其毛发和虹膜色泽逐渐变浅,为黄色或棕黄色,皮肤白。
由于尿液、汗液含有大量苯乳酸而有鼠尿臭味。
随着年龄增长,逐渐表现出智能发育迟缓,以认知发育障碍为主、小头畸形、癫痫发作,也可出现行为、性格等异常,如多动、自残、攻击、自闭症、自卑、忧郁等神经系统表现。
婴儿期还常出现呕吐、湿疹等。
辅助检查1.血苯丙氨酸测定(1)荧光定量法:检测干血滤纸片中Phe 浓度,正常血Phe 浓度<120μmol/L (2mg/dl),血Phe 浓度>120 μmol/L 提示高苯丙氨酸血症。
根据遗传系谱图推断遗传方式的规律
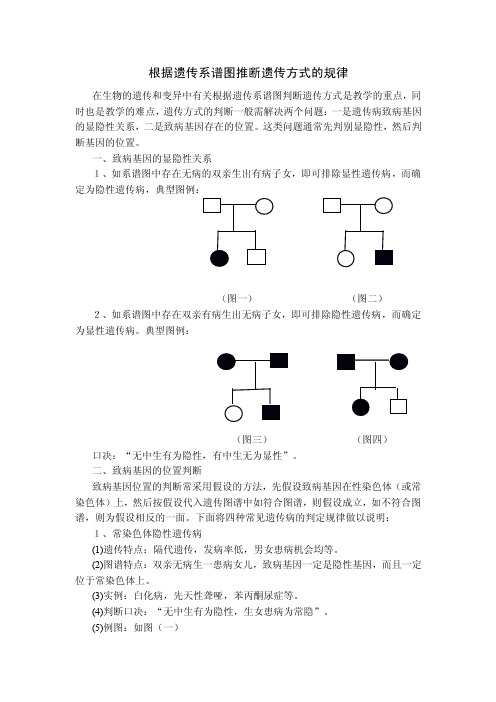
根据遗传系谱图推断遗传方式的规律在生物的遗传和变异中有关根据遗传系谱图判断遗传方式是教学的重点,同时也是教学的难点,遗传方式的判断一般需解决两个问题:一是遗传病致病基因的显隐性关系,二是致病基因存在的位置。
这类问题通常先判别显隐性,然后判断基因的位置。
一、致病基因的显隐性关系1、如系谱图中存在无病的双亲生出有病子女,即可排除显性遗传病,而确定为隐性遗传病,典型图例:(图一)(图二)2、如系谱图中存在双亲有病生出无病子女,即可排除隐性遗传病,而确定为显性遗传病。
典型图例:(图三)(图四)口决:“无中生有为隐性,有中生无为显性”。
二、致病基因的位置判断致病基因位置的判断常采用假设的方法,先假设致病基因在性染色体(或常染色体)上,然后按假设代入遗传图谱中如符合图谱,则假设成立,如不符合图谱,则为假设相反的一面。
下面将四种常见遗传病的判定规律做以说明:1、常染色体隐性遗传病(1)遗传特点:隔代遗传,发病率低,男女患病机会均等。
(2)图谱特点:双亲无病生一患病女儿,致病基因一定是隐性基因,而且一定位于常染色体上。
(3)实例:白化病,先天性聋哑,苯丙酮尿症等。
(4)判断口决:“无中生有为隐性,生女患病为常隐”。
(5)例图:如图(一)2、常染色体上的显性遗传病(1)遗传特点:代代相传,发病率高,男女患病机会均等。
(2)图谱特点:双亲有病生一无病的女儿,致病基因是显性基因,且一定位于常染色体上。
(3)实例:并指、多指、软骨发育不全等。
(4)判断口决:“有中生无为显性,生女正常为常显”。
(5)例图:如图(三)3、伴X隐性遗传病(1)遗传特点:交叉遗传,母病子必病,女病父必病,男性患者多于女性。
(2)实例:红绿色盲,血友病等(3)判断口决:“母病子必病,女病父必病”。
(4)判断方法:否定式判断(5)例图如上图可排除伴X隐性遗传的可能4、伴X显性遗传病(1)遗传特点:连续遗传,交叉遗传,父病女必病,子病母必病,女性患者多于男性。
遗传病口诀

常隐白聋苯色友肌性隐
常显多并软抗D为性显
多裂无高尿异性愚猫叫
注解:1.白:白化病 2.聋:先天性聋哑3.苯:苯丙酮尿症4.色:色盲 5.友:血友病6.肌:进行性肌营养不良7.多:多指症(传说中的六指)8.并:并指9.软:软骨发育不全10.抗D:抗维生素D佝偻病11.多:有多组等位基因控制12.裂:唇腭裂13.无:无脑儿14.高:原发性高血压15.糖:青少年型糖尿病16.异:染色体异常17.猫:猫叫综合症(5号染色体缺失)18.愚:先天愚型(二十一三体综合征)19.性变:性腺发育不良(X型),XXY型,XYY型
无中生有是隐性, 隐性遗传看女病, 女患男正非伴性.
有中生无是显性, 显性遗传看男病, 男正女患非伴性.
伴Y遗传是父传子
1.常染色体显性遗传病:地中海贫血、家族性高胆固醇血症
2.常染色体隐性遗传病:半乳糖血症、黑蒙性痴呆、镰刀型细胞贫血症
3.伴X 染色体隐性遗传病:先天性夜盲症
4.伴Y 染色体遗传病:外耳道多毛症。