荧光原位杂交-更新
荧光原位杂交 综述

荧光原位杂交(FISH)综述摘要本文简单介绍了荧光原位杂交(FISH)技术的一些基础理论知识以及常用操作方法和步骤。
关键词:荧光原位杂交;1.发展荧光原位杂交(fluorescent in situ hybridization,FISH)是一种细胞遗传学技术,可以用来对核酸进行检测和定位。
荧光标记的核酸探针只和具有高度相似性的核酸杂交,可用于染色体上基因的定位,或在分子生态学中用来标记不同分类细菌或古菌中的核糖体RNA[1]。
1969年,Pardue等和John两个研究小组发明了原位杂交技术,放射性标记的DNA 或28s RNA 被杂交到细胞制备物上,通过放射自显影技术(m icroautoradiography, MAR)检测杂交位点,这一技术可以在保持细胞形态完整性的条件下,使核酸序列在细胞内被检测[2]。
2.原理通过特定分子的荧光标记探针在细胞内与染色体上特意的互补核酸序列原位杂交,通过激发杂交探针的荧光来检测信号。
由于荧光燃料收到一定波长的(即激发波长)的光激发后会发射荧光(即发射波长),所以就滤光镜选择合适的激发波长的光,即可显示某一特定的荧光染料,于是就可以直接显示特定细胞核中或染色体上的DNA序列间相互位置关系[2]。
原位杂交的处理:染色体上杂交的位点提供了DNA探针序列的定位信息。
所以应用该方法时,需打开维持染色体DNA双螺旋结构的碱基配对以使其形成单链分子(这称为DNA变性)。
只有这样染色体DNA才能与探针杂交。
变性染色体DNA而不破坏其形态的标准方法是将染色体干燥在玻璃载玻片上,再用甲酰胺处理[1]。
3.关于探针的发展早期原位杂交技术中探针是放射性标记的,但这个方法并不令人满意,因为放射性标记很验证同时满足灵敏度和分辨率这两个原位杂交成功的必要条件。
灵敏度要求放射性标记具有高中辐射能(例如用32P标记),当标记物能量过高时,会因为信号散射导致分辨率过低。
如果使用低辐射能的放射性标记物,如3H可以得到较高的分辨率,但由于灵敏度低而需要长时间曝光,并由此导致背景过高,难以分辨出真正的信号。
荧光原位杂交
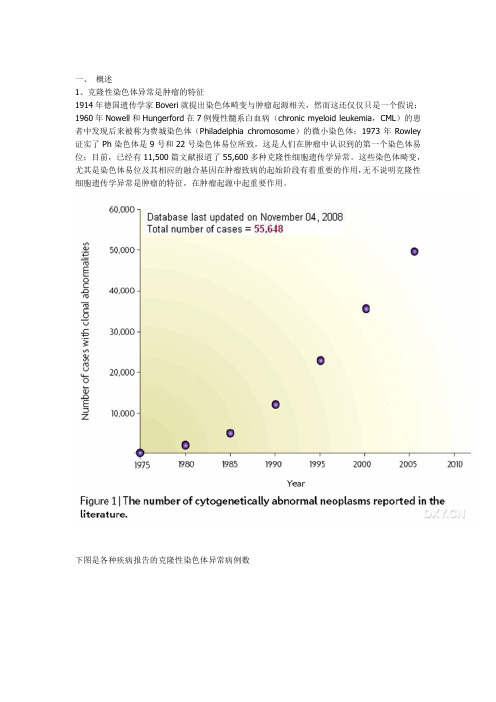
一、概述1、克隆性染色体异常是肿瘤的特征1914年德国遗传学家Boveri就提出染色体畸变与肿瘤起源相关,然而这还仅仅只是一个假说;1960年Nowell和Hungerford在7例慢性髓系白血病(chronic myeloid leukemia,CML)的患者中发现后来被称为费城染色体(Philadelphia chromosome)的微小染色体;1973年Rowley 证实了Ph染色体是9号和22号染色体易位所致,这是人们在肿瘤中认识到的第一个染色体易位;目前,已经有11,500篇文献报道了55,600多种克隆性细胞遗传学异常。
这些染色体畸变,尤其是染色体易位及其相应的融合基因在肿瘤致病的起始阶段有着重要的作用,无不说明克隆性细胞遗传学异常是肿瘤的特征,在肿瘤起源中起重要作用。
下图是各种疾病报告的克隆性染色体异常病例数2、染色体异常的常见类型染色体异常指数目异常和结构异常两类:前者包括整条染色体数目的扩增和缺失;后者包括染色体易位、插入、倒置、区带的缺失或扩增等。
下图是染色体数目异常染色体结构异常3、染色体异常的检测方法染色体异常的识别得益于二十世纪六十年代后发展起来的胰蛋白酶-姬姆萨染色和常规显带技术,使得常规筛查全基因组染色体异常和检测染色体核型改变成为可能。
染色体显带是细胞遗传学分析技术中标准和常用的方法,但耗时且依赖于获得良好的分裂相,还难于分析复杂和隐匿的异常。
PCR或荧光原位杂交(FISH,fluorescent in situ hybridization)对染色体异常的检出依赖于引物或探针与模板的结合,因此较常规显带具有更高的特异性,是高通量检测染色体异常的敏感和特异的方法。
二、荧光原位杂交及其探针1、荧光原位杂交的原理染色体荧光原位杂交始于传统的细胞遗传学和DNA技术的结合,这种结合开创了一门新的学科——分子细胞遗传学。
其基础是Southern blot原理,以半抗原如生物素、地高辛间接标记或以荧光素直接标记的已知核酸分子为探针,探针和靶序列双链DNA变性后杂交,互补的异源单链DNA分子在适宜的温度和离子强度下退火形成稳定的异源双链DNA,通过荧光标记的亲和素或抗地高辛抗体将半抗原显示出来,通过荧光显微镜观察杂交信号。
荧光原位杂交

Capture antibody targeted fluorescence in situ hybridization (CAT-FISH):Dual labeling allows for increased speci ficity in complex samplesJoyce M.Stroot a ,1,Kelly M.Leach a ,Peter G.Stroot b ,2,Daniel V.Lim a ,⁎a University of South Florida,Department of Cell Biology,Microbiology,and Molecular Biology,4202E.Fowler Ave.ISA2015,Tampa,FL 33620,USA bUniversity of South Florida,Department of Civil and Environmental Engineering,4202E.Fowler Ave.ENB118,Tampa,FL 33620,USAa b s t r a c ta r t i c l e i n f o Article history:Received 3November 2011Received in revised form 13December 2011Accepted 13December 2011Available online 22December 2011Keywords:Cytometric bead array Escherichia coli O157:H7Fluorescence in situ hybridization ImmunoassayStaphylococcus aureusPathogen detection using biosensors is commonly limited due to the need for sensitivity and speci ficity in detecting targets within mixed populations.These issues were addressed through development of a dual la-beling method that allows for both liquid-phase fluorescence in situ hybridization (FISH)and capture anti-body targeted detection (CAT-FISH).CAT-FISH was developed using Escherichia coli O157:H7and Staphylococcus aureus as representative bacteria,and processing techniques were evaluated with regard to FISH intensities and antibody recognition.The alternative fixative solution,methacarn,proved to be superior to standard solid-phase paraformaldehyde fixation procedures,allowing both FISH labeling and antibody rec-ognition.CAT-FISH treated cells were successfully labeled with FISH probes,captured by immunomagnetic separation using fluorescent cytometric array beads,and detected using a cytometric array biosensor.CAT-FISH treated cells were detectable with LODs comparable to the standard antibody-based technique,(~103cells/ml in PBS),and the technique was also successfully applied to two complex matrices.Although immunomagnetic capture and detection using cytometric arrays were demonstrated,CAT-FISH is readily ap-plicable to any antibody-based fluorescence detection platform,and further optimization for sensitivity is possible via inclusion of fluorescently tagged antibodies.Since the con fidence level needed for positive iden-ti fication of a detected target is often paramount,CAT-FISH was developed to allow two separate levels of speci ficity,namely nucleic acid and protein signatures.With proper selection of FISH probes and capture an-tibodies,CAT-FISH may be used to provide rapid detection of target pathogens from within complex matrices with high levels of con fidence.©2011Elsevier B.V.All rights reserved.1.IntroductionPathogen detection using biosensors is commonly limited due to the need for both sensitivity and speci ficity in detecting targets within the mixed populations present in complex samples (Lim et al.,2005).The relatively high detection limits inherent in most systems are in fluenced by many factors,including low target concentrations,poor capture ef fi-ciencies,non-target detection (false positives/negatives)and interfer-ence by organic/inorganic constituents,all of which precludes adequate detection (Simpson-Stroot et al.,2008).Additionally,for those systems using immunochemistry for target capture or reporting (e.g.antibodysandwich assays),non-target cross-reactivity issues can be problematic,and adequately speci fic antibodies are often not available.Some of these limitations may be overcome by combining two dif-ferent molecular signature techniques,which would bestow added con fidence for identifying the presence of targeted pathogens —in particular,the use of speci fically-targeted fluorescently-labeled 16S rRNA gene oligonucleotide probes in conjunction with speci fically-targeted antibodies.The dual level speci ficity (nucleic acid and pro-tein)allows two levels of accuracy for detection and/or con firmation,as well as addressing cross-reactivity.For example,if the antibody available for a given target cross-reacts with other bacteria (related or otherwise),it could still be used for antibody capture-dependent biosensors,as long as a labeled nucleic acid speci fic probe is used to generate the signal (as opposed to a labeled detector antibody).This probe would only provide fluorescent signal to the appropriate target.Thus,the binding of unlabeled non-target cells to the antibodies be-comes a null issue as no signal is generated.The use of fluorescence in situ hybridization (FISH)to phylogenet-ically identify microorganisms without cultivation based on either 16S or 23S rRNA has become a mainstay of microbial ecology since its introduction (DeLong et al.,1989;Amann et al.,1990a,b;Amann,Journal of Microbiological Methods 88(2012)275–284⁎Corresponding author at:University of South Florida,Department of CMMB,4202E.Fowler Ave.,ISA2015,Tampa,FL 33620-7115,USA.Tel.:+18139741618;fax:+18139059940.E-mail addresses:jstroot@ (J.M.Stroot),kmleach@ (K.M.Leach),pstroot@ (P.G.Stroot),lim@ (D.V.Lim).1Present Address:Ciris Energy Inc.,9155E.Nichols Ave.Ste.200,Centennial,CO 80112,USA.2Present Address:11570Crow Hill Dr.Parker,CO 80134,USA.0167-7012/$–see front matter ©2011Elsevier B.V.All rights reserved.doi:10.1016/j.mimet.2011.12.009Contents lists available at SciVerse ScienceDirectJournal of Microbiological Methodsj o u r n a l h o m e p a g e :w w w.e l s e vi e r.c o m/l o c a t e /j m i c m e t h1995;Amann et al.,2001;Wagner et al.,2003;Daims et al.,2005). FISH has also been reported as a rapid method for pathogen identifi-cation in clinical and food settings(Kempf et al.,2000;Hartmann et al.,2005;Kempf et al.,2005;Peters et al.,2006;Wellinghausen et al.,2006;Bisha and Brehm-Stecher,2009a,b).Although the predomi-nant FISH application has been to study microbial community struc-ture and spatial arrangements on solid supports,some applications have explored its usefulness forflow cytometric analyses(Amann et al.,1990a,b;Wallner et al.,1993,1997;Fuchs et al.,1998;Hartmann et al.,2005;Kempf et al.,2005).This adaptation to a liquid phase pro-cessing forflow cytometry lends itself to facilitating biosensor appli-cations,provided that conditions allowing for both probe and antibody recognition are met.Traditionally,samples to be processed by FISH arefixed with para-formaldehyde(PFA)to stabilize and preserve them(Daims et al., 2005).PFA acts as a strengthening agent on the membranes of Gram-negative bacteria by cross-linking proteins to prevent lysis dur-ing hybridization,but can make Gram-positive bacteria highly resis-tant to probe uptake(Leong,1994;Daims et al.,2005).Additionally, this cross-linking activity,while giving stability and excellent condi-tions for FISH,can severely inhibit any subsequent immunochemistry. To circumvent these problems,combined bacterial applications of FISH and immunostaining have typically involved extensive antibody incubation times or involved processing steps to overcome thefixa-tive effects(Aβmus et al.,1997;Li et al.,1997;Ramage et al.,1998; Oerther et al.,1999),limiting their utility for rapid and high-throughput testing situations.As formaldehyde and its derivatives are well known in the histo-pathology community to inhibit molecular analyses(e.g.immunos-taining,immunohistochemistry or nucleic acid analysis),alternative tissuefixatives have been explored that are more conducive to down-stream processing(Baumgärtner et al.,1988;Leong,1994;Shibutani et al.,2000;Srinivasan et al.,2002;Cox et al.,2006).Methacarn solu-tion has been found to be a non-cross-linking protein-precipitating fixative that does not appear to affect polynucleotide or protein anal-ysis offixed tissues and usually will give superior immunohistochem-ical results(Shibutani and Uneyama,2002).This success with tissues suggests that methacarn solution may also be successful withfixation of bacterial cells and lend itself to facilitating the use of FISH in com-bination with immunolabeling for biosensor detection.In the work described herein,we demonstrate a modified liquid FISH processing method used in conjunction with capture antibody targeted detection(CAT-FISH)to increase the specificity for biosensor assays.Detection of pathogens in pure cultures and seeded matrices was demonstrated on a cytometric bead array biosensor,using bead-bound capture antibodies with FISH labeled cells.Since the ap-plications of both FISH and immunochemistry have been well estab-lished for use with complex sample matrices,this method should be easily adapted to other bacteria and biosensor platforms.The use of FISH in conjunction with antibody based biosensor assays for patho-gen detection has not been previously reported.2.Materials and methods2.1.MicroorganismsEscherichia coli O157:H7(ATCC35150)and Staphylococcus aureus (ATCC25923)were cultured at37°C with shaking in brain heart in-fusion broth(BHI)(Difco,Becton Dickinson,Franklin Lakes,NJ). Cells were cultured for~18h prior to experiments and then diluted 1:100in fresh BHI and incubated as described.Cells were harvested at0.8O.D.(600nm)and washed once with1×phosphate buffered saline(PBS−11.9mM phosphate buffer,137mM sodium chloride, 2.7mM potassium chloride,pH7.4[Fisher BioReagent,Suwanee, GA]).Cell pellets were thenfixed immediately(PBS samples)or dilut-ed into complex matrix prior tofixation.2.2.FixationHarvested bacteria werefixed by one of two methods:Control cells werefixed with4%ice-cold paraformaldehyde(PFA)for2h at 4°C,collected by centrifugation(9000×g)for90s,decanted and resuspended in50%(v/v)ethanol in1×PBS according to standard FISH protocols(Daims et al.,2005).Test cells werefixed with metha-carn solution(6:3:1parts absolute methanol,chloroform,and glacial acetic acid)made fresh daily(Leong,1994).All manipulations involv-ing the methacarn solution were performed in a chemical hood.Cell pellets were resuspended in methacarn,incubated at room tempera-ture(22°C)for10min,collected by centrifugation(9000×g)for90s, decanted,and washed once in80%(v/v)ethanol and once in100% ethanol.Cell pellets were dried in the dark via rotation for20min at 46°C to drive off remaining ethanol(Enviro-Genie,Scientific Indus-tries,Inc.,Bohemia,NY).Samples containing spinach rinse(spiked with E.coli O157:H7or un-amended)were processed in the same manner.For Staphylococcus samples,prior to alcohol dehydration steps,cells were treated with lytic agents as follows:after centrifuga-tion,cells were washed once in10mM Tris HCl pH8.0,centrifuged and resuspended in lysozyme(1mg/ml in10mM Tris HCl pH.8.0) and incubated15min at30°C;then cells were collected as described and resuspended in either lysostaphin(10μg/ml in10mM Tris HCl pH8.0)and incubated5min at30°C or nisin(0.1g of2.5%powder in10ml0.02N HCl pH2.0)diluted into PBS forfinal concentration of25μg/ml).2.3.Liquidfluorescence in situ hybridization(LQ-FISH)Fluorescently-labeled oligonucleotide hybridization probes target-ing the16S rRNA for the domain Eubacteria(EUB338;5′gctgcctcccgtag-gagt3′)and species S.aureus(SAU349;5′gaagcaagcttctcgtccg3′)were obtained from Molecular Probes(Invitrogen,Carlsbad,CA)and labeled with either Alexa Fluor532or Cy3at the5′end(Loy et al.,2007).Alexa Fluor532labeled probe was used for cytometric bead array assays,and Cy3labeled probe was used for optimization assays,image capture and image analysis.The probes were diluted to50ng/μl with dH2O,and stored in100-μl aliquots at−20°C in the dark.Fixed and dried sample pellets(pure cultures or spiked matrix)were resuspended in200μl of hybridization buffer containing the labeled probe at afinal concentra-tion of5ng/μl.The hybridization buffer also contained20%(v/v)form-amide,0.9M NaCl,0.1%SDS and100mM Tris HCl[pH7.0](Daims et al., 2005).Hybridization was conducted in the dark for1h in a46°C water bath.Subsequently,samples were centrifuged(9000×g)for90s,the supernatant was removed,and cells were resuspended in500μl of pre-warmed washing buffer followed by10min incubation in a48°C water bath in the dark.The washing buffer contained215mM NaCl, 20mM Tris HCl[pH7.0]and5mM EDTA(Daims et al.,2005). After washing,samples were centrifuged and the supernatant removed. Treated pellets were resuspended in PBS for downstream processing (microscopy,antibody labeling,or immunomagnetic capture and cytometric analysis).2.4.Standardfluorescence in situ hybridizationVariations in oligonucleotide conferredfluorescence intensity based uponfixative affect were measured.Cellsfixed by either PFA or methacarn were placed on10-well heavy Teflon coated micro-scope slides(Cel-Line Associates,New Field,NJ)and processed by standard FISH protocols(Amann et al.,1990a,b;Oerther et al.,2000) using the Cy3-labeled EUB338probe.Lytic agents were applied to the Staphylococcus samples as described in Section2.2.The hybridiza-tion step was1h and the washing step was30min.Cells were coun-terstained with4′,6-diamidino-2-phenylindole(DAPI)at a concentration of1μg/ml for1min,rinsed with dH2O,air dried,and276J.M.Stroot et al./Journal of Microbiological Methods88(2012)275–284mounted with Cargille immersion oil(Type FF,Cedar Grove,NJ)and a cover slip.2.5.Antibody binding assaysFixed cells were used in adsorption enzyme linked immunosor-bent assays(ELISA)to compare antibody recognition after the differ-entfixation techniques.Bacteria werefixed with either PFA or methacarn as described above and split,with half used as controls and half processed alike through the entire liquid FISH procedure. Additional controls consisted of untreated cells that were harvested and stored on ice in PBS from the same initial test cultures.After treatments,cells were enumerated by direct microscopic count,nor-malized to1×108cells/ml and serially diluted to103cells/ml in PBS. Preliminary experiments indicated that cell concentrations below 102cells/ml did not produce measurable signal and were omitted from further tests(data not shown).Cells were then applied in trip-licate to an ELISA plate,incubated at4°C~18h and processed using a QuantaBlu Fluorogenic Peroxidase kit(PIERCE,Rockford,IL) according to manufacturer's instructions with the following excep-tions:volumes for each step were normalized to100μl and incuba-tion times were reduced to30min each.Reporter antibodies included affinity purified peroxidase labeled polyclonal goat anti-E. coli O157:H7[1μg/ml](KPL,Gaithersburg,MD),purified rabbit anti-S.aureus[10μg/ml](AbD Serotec,Raleigh,NC)and affinity pu-rified peroxidase labeled goat anti-rabbit[1μg/ml](KPL).ELISA plates were washed using an EL x50auto-strip washer(Bio-Tek In-struments Inc.,Winooski,VT)and end product detection performed on a SpectraMax GeminiXS Fluorometer(Molecular Devices,Silicon Valley,CA).Results are reported as signal to noise(S/N)ratios for test samples versus PBS,where an S/N ratio above3indicates a pos-itive detection result.2.6.Simultaneous FISH and reporter antibody labelingE.coli O157:H7and S.aureus samples were individuallyfixed in methacarn and processed according to the liquid FISH method de-scribed in Section2.3either with or without Cy3-labeled EUB338 probe.Treated cells were resuspended in500μl PBS containing either 4μg/ml goat anti-E.coli O157:H7(KPL-for E.coli O157:H7samples) or4μg/ml biotin conjugated rabbit anti-S.aureus(AbD Serotec—for S.aureus samples),and incubated at room temperature for30min in the dark with end over end rotation(24rpm).The cells were re-covered by centrifugation(9000×g for90s)and washed once in 500μl PBS with5min end over end rotation.E.coli and S.aureus sam-ples were then resuspended in500μl PBS containing2μg/ml Cy2-labeled donkey anti-goat(Jackson ImmunoResearch Laboratories, Inc.,West Grove,PA)or2μg/ml streptavidin conjugatedfluorescein isothiocyanate(FITC)(Jackson ImmunoResearch),respectively.Cells were incubated at room temperature for30min in the dark with end over end rotation,washed,resuspended in100μl PBS,and visual-ized using an epifluorescent microscope.2.7.Cytometric assaysA method was developed to incorporate immunomagnetic separa-tion(IMS)with cytometric bead arrays for direct detection of FISH la-beled cells(IMS-CAT-FISH)using a specializedflow cytometer.This method is a modification to standard cytometric bead array(CBA)as-says,which rely on a labeled detector antibody for target detection. MagPlexC magnetic microbeads(6.5±0.2μm,fluorescence region 33,Luminex,Austin,TX)were used for all assays,and for all manipu-lations beads were separated from liquid solutions using the3-in-1 Magnetic Particle Separator(PureBiotech,LLC,Middlesex,NJ).Mag-netic microspheres were coupled to goat anti-E.coli O157:H7anti-body(KPL)or rabbit anti-S.aureus(AbD Serotec)using the amine coupling kit(BioRad,Hercules,CA),as per the manufacturer's instruc-tions and stored at4°C until used.Coupled magnetic microspheres were used to capture target cells from sample suspensions following the LQ-FISH procedure.Antibody-coupled microbeads(1000per sample)were added to1ml of sample and briefly mixed by vortex action on low setting.Mixed samples were centrifuged(6000×g,60s)and target allowed to bind during a5min stationary incubation at22°C.Samples were briefly mixed after incubation and beads were separated from the supernatant.Re-covered beads were washed twice with500μl PBS containing0.05% Tween20(PBST)in the dark using5min rotations.After washing steps,beads were separated from the sample supernatant and resus-pended in100μl PBST.Beads were placed into96-well round-bottom plates,and analyzed using the Bio-Plex200reader(BioRad),as per the manufacturer's instructions using the high PMT setting.Fluores-cence signals were expressed as the meanfluorescence intensity of 100beads per well.Positive signals were determined using signal above limit of detec-tion(SALOD)values.SALODs were calculated by taking the average sample signal and subtracting the limit of detection(LOD)value. The LOD value was calculated by averaging sample blanks and adding three times the standard deviation of the sample blanks.All experi-ments were performed in duplicate or triplicate.2.8.Simulation of complex testing situations2.8.1.Spinach rinse matrix preparationSpinach rinse was generated to simulate afield wash sample as E.coli O157:H7contamination is a concern in commercial produce products.Fresh bundled spinach was purchased from a local wholesale market(Tampa,FL)and processed within one day.Spin-ach rinse was generated and stored at−20°C until used.Rinses were tested for background E.coli O157:H7contamination accord-ing to established FDA-BAM procedures(Feng et al.,1998),and de-termined to be devoid of endogenous E.coli O157:H7prior to initiation of experiments.Serially diluted samples were plated on BHI agar using the spread plate method to determine microbial background loads.Prior to IMS-CAT-FISH,E.coli O157:H7was diluted into spinach rinse,mixed1:1with2×modified buffered peptone water with py-ruvate(mBPWp),(Feng et al.,1998),and enriched overnight at 42°C,statically.After enrichment,spiked and un-spiked samples were plated onto CT-SMAC agar to verify growth in spiked samples and absence of target in controls.Before sampling,enriched samples were mixed briefly and allowed to stand for1min to allow for sand and large particulates to settle out.Samples for analysis were collected from the resultant supernatant,and processed for IMS-CAT-FISH,as described.Assay samples included bead only controls, un-spiked PBS blanks,spiked PBS samples,and un-spiked spinach rinse blanks to evaluate possible background interference from non-specific binding.2.8.2.Blood culturesCitrated sheep blood(Fisher Scientific)was used as a surrogate for human blood.Blood samples were spiked with S.aureus at con-centrations of~2CFU/ml(verified by plate counts on BHI agar),and diluted1:5into BACTEC-derived blood culture media,which was prepared in-house and contained3%soybean-casein digest,0.3% yeast extract,0.01%beef extract,0.1%sucrose,0.0005%hemin, 0.00005%menadione,0.001%Pyridoxal HCL,0.04%sodium bicar-bonate,and0.035%sodium polyanetholsulfonate(all w/v).Samples were enriched overnight at37°C,with aeration,and plated on BHI agar to verify bacterial growth in spiked samples,and absence of bacteria in non-spiked samples.500μl of the blood culture sample was used for each assay.The sample was mixed with500μl water and incubated for5min at room temperature to permit lysis of277J.M.Stroot et al./Journal of Microbiological Methods88(2012)275–284remaining erythroid cells.Samples were then centrifuged to pellet bacteria,re-suspended in1ml PBS,and processed according to the IMS-CAT-FISH procedure.Prior to IMS capture,samples were diluted1:100in order to dilute particulates resulting from remain-ing blood components.2.9.Microscopic imaging and data analysisSamples for image analysis were placed on10-well microscope slides(Cel-Line)and either observed under wet mount or air-dried. Air dried cells were mounted with Cargille immersion oil(Type FF) and a cover slip.Oligonucleotide and/or antibody conferredfluores-cence were visualized with upright epifluorescence microscopes,ei-ther a Leitz DiaPlan(Heerbrugg,Switzerland)or an Olympus BX60F (Center Valley,PA).Digital images were captured using Spot-FLEX CCD cameras and image modifications(for publication purposes only)were performed using Spot Software4.6(Diagnostic Instru-ments,Inc.,Sterling Heights,MI).Image modifications consisted of cropping,scale bar calibrations and pixel multiplicative brightness adjustments.Brightness adjustments were scaled up a factor of2or 3dependent upon image series,however within each series the mul-tiplicative factor was the same,so that relative intensities were com-parable.All data analysis was performed on raw images without modifications.Fluorescent images were evaluated with the daime 1.3.1software package(Daims et al.,2006).Measure objects analysis calculated the number of cells detected,as well as the meanfluores-cent intensity and standard deviations for each cell.SigmaPlot®10 (Systat Software,Inc.,San Jose,CA)was used for graphic analysis.Stu-dent's t test was used to determine if significant differences occurred betweenfixative types.3.Results3.1.Optimization of processing conditionsPreliminary evaluations of multiplefixatives and conditions,in-cluding standard FISHfixation with PFA,did not facilitate sufficient antigen–antibody binding as required for target capture and detec-tion on biosensors(data not shown).Experiments attempting to as-certain antigen–antibody binding capabilities for stored PFAfixed E. coli cells were unsuccessful in producing any antibody binding (data not shown).Thus all subsequent experiments used“fresh”fixed cells for evaluation purposes,in that cells were processed im-mediately afterfixation steps without holding or storage.Methacarn fixation was the most promising candidate for bothfluorescence and antibody binding and was subsequently optimized for use in the LQ FISH protocol.3.2.Influence offixative uponfluorescent signalE.coli O157:H7and S.aureus werefixed in either methacarn or PFA to determine if methacarnfixation produced cells capable of binding probe and emittingfluorescence signals comparable to stan-dard protocols.Cells from bothfixative treatments(methacarn and PFA)were mounted on slides and subjected to standard FISH.In addi-tion,cells were processed by the LQ-FISH method using methacarn fixation,and spotted onto separate slides(Fig.1).Relativefluores-cence intensity means for E.coli O157:H7cells(200per treatment) were:61±12for PFA,68±11for methacarn and58±10for LQ-FISH.Analyses indicated that there were no significant differences be-tween the PFA and LQ-FISH treatments,but the methacarn treated cells were significantly brighter(p≤0.0004)than both PFA and LQ-FISH cells.Relativefluorescence intensity means for S.aureus cells (150per treatment)were:48±14for PFA,56±18for methacarn and100±28for LQ-FISH.Analyses indicated that the PFA treated cells were significantly dimmer(p≤0.003)than both methacarn and LQ-FISH cells,and LQ-FISH treated cells were significantly brighter than the slide based methacarn treated cells(p≤0.003). 3.3.Influence offixative upon antibody bindingAdsorbent ELISAs were used to evaluate PFA and methacarnfix-ation effects.In addition,fixed cells were subjected to further hy-bridization conditions(LQ FISH)to determine the effect of hybridization treatments on antibody binding for E.coli O157:H7 and S.aureus(Fig.2).Antibody binding was significantly reduced for PFAfixed E.coli as compared to control(non-fixed)and metha-carnfixed cells for all concentrations below108cells/ml(Fig.2A). Positive S/N ratios were observed for methacarn,control,and PFA at104,105,and106cells/ml,respectively.Methacarn treatment achieved better binding than controls at concentrations below 105cells/ml and showed no difference at higher concentrations. After LQ-FISH hybridization treatments,methacarn treated cells produced higher S/N ratios than PFA treated cells;however,the minimum concentration producing a positive signal was one log higher as compared to non-hybridized cells(Fig.2B).No change in minimum concentration was observed for the PFA treated cells after hybridization.Antibody binding for S.aureus produced no sig-nificant differences between controls and methacarn or PFAfixed cells,and positive S/N ratios were achieved at106cells/ml for all conditions(Fig.2C).After hybridization,the methacarnfixed cells produced slightly higher S/N ratios,but were not significantly dif-ferent from PFA samples(Fig.2D).3.4.Simultaneous visualization of oligonucleotide and antibodyfluorescenceBoth labeling methods were used in conjunction to examine label-ing coverage after methacarnfixation with LQ-FISH hybridization.E. coli O157:H7and S.aureus were simultaneously labeled with rRNA probes and antibodies and generated signals that were sufficient for microscopic visualization(Fig.3).Appropriate un-labeled controls were included(±probe and/or antibody)and verified(data not shown).Visual examination of E.coli cells indicated that they were uniformly labeled with both CY3FISH probe and CY2antibodies,pro-viding ample coverage for dual level specificity detection.S.aureus cells also exhibited dual labeling,but the signal intensities appeared to be more variable for bothfluorescent markers.3.5.Immunomagnetic cytometric bead array detectionA cytometric bead array biosensor was selected as an example platform to demonstrate the utility of the dual specificity target de-tection.Cells were labeled using LQ-FISH and captured by IMS using antibody-coupled magnetic cytometric array microspheres (IMS-CAT-FISH).Bead-associatedfluorescence signal was then deter-mined using the cytometric array reader.Initially,limit of detection assays for E.coli O157:H7and S.aureus were performed in PBS, and microscopic evaluation of E.coli O157:H7was used to visually verify labeling and capture.Utility of the method was subsequently verified using a more complicated testing matrix appropriate for each organism.In addition,S.aureus limit of detection assays were performed using either species specific lytic treatments(lysosta-phin)or general gram-positive lytic treatment(nisin),incorporated into the LQ-FISH procedure.3.5.1.E.coli O157:H7detection assaysWe previously reported that standard cytometric bead array(IMS-CBA)assays using labeled detector antibodies are useful for rapid de-tection of E.coli O157:H7in both PBS and a more complex testing sce-nario(raw spinach testing via enrichment of rinse samples)(Leach et al.,2010).However,in spinach rinse enrichment samples,some non-278J.M.Stroot et al./Journal of Microbiological Methods88(2012)275–284speci fic background signals were encountered,suggesting the need for a more speci fic detection procedure (Leach et al.,2010).Toward this end,CAT-FISH was applied.Microscopic evaluation was first conducted to evaluate target capture and labeling for both the standard IMS-CBA assay (antibody label)and the IMS-CAT-FISH method (FISH label).All samples (bead only controls,un-spiked PBS blanks,spiked PBS samples,and un-spiked spinach rinse blanks)were spotted onto slides and examined (Fig.4).Beads incubated without target or reporter pro-duced no detectable signal in either assay (not shown).Detector-associated signal for PBS blanks was absent from CAT-FISH treat-ments (i.e.,no labeled cells present,not shown),but a dim signal was visible after processing according to standard array assay method (Fig.4A).Beads incubated with control cells produced highly visible signal for both treatments.E.coli detected with the standard assay produced images with visible halos surrounding the surface of the captured cells,corresponding to the binding of la-beled detector antibody to bead-captured beled cells were easily distinguishable from the bead surfaces (Fig.4C).E.coli cells detected with CAT-FISH produced images of solidly labeled cells bound to the surface of the beads (Fig.4D).Non-spiked spinach blanks treated with CAT-FISH produced no detectable signal (not shown).Interestingly,there was observable signal on beads after the standard assay was performed on the un-spiked rinse.Fluores-cence images displayed relatively large patches of labeled non-target particulate material attached to the cytometric array beads (Fig.4B),thus demonstrating non-speci fic binding of spinach debris to the beads and the reporter antibody.As microscopic evaluation indicated successful labeling and cap-ture,a cytometric array biosensor (BioPlex 200)was used to evaluate target signals obtained using the IMS-CAT-FISH assay.Detection was sporadically observed at 103cells/ml and routinely observedatparison of relative fluorescence intensities for E.coli O157and S.aureus treated with different fixatives.All cells were labeled with EUB338Cy3rRNA FISH probe.Cells fixed with either PFA or methacarn were fixed prior to mounting on slides for FISH processing.Cells treated via LQ-FISH were mounted on slides after the process was complete.A and D)PFA treatment;B and E)methacarn treatment;C and F)LQ-FISH treatment.Scale bar is 5μm.279J.M.Stroot et al./Journal of Microbiological Methods 88(2012)275–284。
荧光原位杂交 综述
荧光原位杂交(FISH)综述摘要本文简单介绍了荧光原位杂交(FISH)技术的一些基础理论知识以及常用操作方法和步骤。
关键词:荧光原位杂交;1.发展荧光原位杂交(fluorescent in situ hybridization,FISH)是一种细胞遗传学技术,可以用来对核酸进行检测和定位。
荧光标记的核酸探针只和具有高度相似性的核酸杂交,可用于染色体上基因的定位,或在分子生态学中用来标记不同分类细菌或古菌中的核糖体RNA[1]。
1969年,Pardue等和John两个研究小组发明了原位杂交技术,放射性标记的DNA 或28s RNA 被杂交到细胞制备物上,通过放射自显影技术(m icroautoradiography, MAR)检测杂交位点,这一技术可以在保持细胞形态完整性的条件下,使核酸序列在细胞内被检测[2]。
2.原理通过特定分子的荧光标记探针在细胞内与染色体上特意的互补核酸序列原位杂交,通过激发杂交探针的荧光来检测信号。
由于荧光燃料收到一定波长的(即激发波长)的光激发后会发射荧光(即发射波长),所以就滤光镜选择合适的激发波长的光,即可显示某一特定的荧光染料,于是就可以直接显示特定细胞核中或染色体上的DNA序列间相互位置关系[2]。
原位杂交的处理:染色体上杂交的位点提供了DNA探针序列的定位信息。
所以应用该方法时,需打开维持染色体DNA双螺旋结构的碱基配对以使其形成单链分子(这称为DNA变性)。
只有这样染色体DNA才能与探针杂交。
变性染色体DNA而不破坏其形态的标准方法是将染色体干燥在玻璃载玻片上,再用甲酰胺处理[1]。
3.关于探针的发展早期原位杂交技术中探针是放射性标记的,但这个方法并不令人满意,因为放射性标记很验证同时满足灵敏度和分辨率这两个原位杂交成功的必要条件。
灵敏度要求放射性标记具有高中辐射能(例如用32P标记),当标记物能量过高时,会因为信号散射导致分辨率过低。
如果使用低辐射能的放射性标记物,如3H可以得到较高的分辨率,但由于灵敏度低而需要长时间曝光,并由此导致背景过高,难以分辨出真正的信号。
荧光原位杂交更新

常用探针类型 — 重复序列探针
重复序列探针的结合位点是基因组中多拷 贝的短重复碱基对序列(例如着丝粒和端 粒探针)所在的染色体区域。 着丝粒常常是A-T丰富区,而端粒已知含有 TTAGGG序列。由于重复序列相对容易制 备和分辨率较高(0.5Mb),这种FISH被 普遍用于筛查一般的染色体非整倍体、亚 端粒缺失和标记染色体。
杂交流程
a.两要素:
探针和靶序列 b.标记探针
直接标记和间接标记
c.变性 探针和靶序列成为单链 d.混合杂交 e.间接标记探针需连接 荧光基团的步骤
FISH
荧光标记的DNA探针 (200-500bp)
荧光原位杂交特点
与其它原位杂交技术相比,FISH的优点:
①经济安全,快速方便; ②敏感性高,特异性强,背景低; ③宜于多靶杂交,对同一标本同时进行多个探针杂 交,不同的探针可以显示不同的荧光颜色; ④使用G带玻片可作出回顾性分析;
结合技术相关检测项目
染色体病快速产前诊断(FISH、qPCR、 BoBs) 产前单基因病诊断 SNP Array 检测 某些代谢病的产前诊断
项目意义
较羊水染色体分析,FISH技术可以确定13、18、21、X、Y的染色体 数目信息,缓解唐筛阳性患者或高龄孕妇精神负担;缓解医生核型分 析工作压力,能够有针对性的对样本进行处理。
工作基础
探针 商品化探针 原位杂交仪 Luminex xMAP 液相芯片检测平台
工作基础
工作人员 1)项目负责人 刘红卫,男,妇产科实验室技术负责人,毕业 于四川大学生物工程系遗传学专业,临床遗传优 生专业副主任医师,丰富的技术操作和管理能力。 2)项目组成员 赵少志,男,检验师,四川大学华西医院医学 遗传学硕士,中国遗传医学中心、医学遗传学国 家培训中心学员。
2024荧光原位杂交技术在血液肿瘤中的应用规范(全文)

2024荧光原位杂交技术在血液肿瘤中的应用规范(全文)荧光原位杂交(FISH)技术是基于碱基互补配对原则,利用荧光标记的核酸探针,对待测标本DNA序列进行定位、定性和定量分析的技术,是遗传学异常的主要检测手段之一。
相较于核型分析,FISH技术具有快速、灵敏度高及特异性强的优势,是血液肿瘤诊断和预后判定的重要手段。
为进一步规范FISH技术在血液肿瘤中的应用,中国抗癌协会血液病转化医学专业委员会、中国老年医学学会病理学分会及中华医学会血液学分会组织国内血液学、病理学和检验学专家,制定了FISH 在血液肿瘤中的应用规范。
1 FISH在血液肿瘤诊疗中的应用价值1.1 诊断分型遗传学改变是血液肿瘤诊断分型的主要依据。
急性白血病(AL)、慢性粒细胞白血病(CML)、伴嗜酸粒细胞增多和酪氨酸激酶基因融合的髓系/淋系肿瘤(MLN-TK)、骨髓增生异常综合征(MDS)、大B 细胞淋巴瘤、滤泡淋巴瘤(FL)、套细胞淋巴瘤(MCL)、伯基特淋巴瘤(BL)、边缘区淋巴瘤(MZL)、间变大细胞淋巴瘤(ALCL)、T幼淋巴细胞白血病(T-PLL)等均需要借助FISH检测关键的遗传学异常才能精准分型。
1.2 预后分层目前针对急性髓系白血病(AML)、急性淋巴细胞白血病(ALL)、MDS、慢性淋巴细胞白血病(CLL)、原发性骨髓纤维化(PMF)、多发性骨髓瘤(MM)等血液肿瘤,世界卫生组织(WHO)、美国国立综合癌症网络(NCCN)指南已提出了基于遗传学异常的预后分层/评分体系。
FISH检测在血液肿瘤的预后评估中发挥着不可替代的作用。
1.3 指导治疗CML患者中费城染色体(Ph染色体)或BCR::ABL1融合基因的发现开启了酪氨酸激酶抑制剂(TKI)靶向治疗的新时代。
此外,针对PML::RARA融合基因或其他RARA重排的全反式维甲酸和砷剂、ABL信号通路(ABL1、ABL2、PDGFRA、PDGFRB重排)的TKI药物、JAK-STAT信号通路(CRLF2、JAK1/2/3重排)的JAK抑制剂、FLT3重排的FLT3抑制剂、ALK重排的ALK抑制剂等均已正式应用于临床治疗或处于临床试验阶段。
染色体荧光原位杂交技术

1.2 探针和染色体的杂交
• 染色体原位杂交:标记后的探针经变性后.加于变性的 中期染色体分裂相标本上。于37摄氏度、50%的甲酰胺 2XSSC条件下进行杂交.最适的杂交条件主要取决于探 针与靶基因的性质。对于单拷贝基因的杂交,特别是较 长的来自基因组的序列,一般在杂交之前应进行预复性 过程,标记的探针与过量的基因组DNA或Cot-1重复序 列一起孵育1小时,使探针中非特异的重复序列被封闭。 从而抑制该部分探针与染色体之间发生非特异性杂交, 上述过程又称为抑制性染色体原位杂交。经预复性之后 已标记好的变性探针(200一500bP)再与变性的染色体在 含有甲酰胺,盐和醋酸葡聚糖的杂交缓冲液中杂交过夜。 接着是柔和的洗涤,以去除未杂交或错配对的探针。
• (2)间接标记法:常用的间接法是将一些 类似于半抗原(hapten)的标记分子掺入探针 分子中。用的较多的试剂有生物素,地谷新 (digoxigenin),二硝基苯(dinitrophenyl, DNP),氨基乙酰茐(aminoacetylfuorene, AAF),汞和磺酸盐(sulfonate)。就掺入方式 来说,生物素,地谷新等是以核苷酸衍生物 的形式掺入的,可用切口平移法来进行。现 在更多的人开始用随机引物法。用这两种方 法标记好的探针大小在200一500bp范围内, 是用于杂交的最佳大小。另外,也可以在已 知顺序的两个引物之间用PCR法来扩增,或 从合适的载体上通过RNA转录而来。
• 荧光标记的染色体原位杂交技术提供了一种快速而有 效的手段,将DNA片段和特定的真核生物细胞的染色 体区带联系了起来,并将这些DNA片段排序,这是研 究DNA顺序在染色体上位置的最直接的方法。最早, 人们根据Gall和Pardue在1969年的工作,于70年代, 建立起了同位素原位杂交技术,但当时只是用于DNA 顺序在多线染色体上的定位或是重复顺序在中期染色 体上的定位。1981,Gerhard和Harper等首先证明有 可能将从个体基因中得到的单拷贝顺序(SCP,single copy probe)通过同位素原位杂交技术定位到中期染色 体上。在此基础上,80年代初起,荧光标记的原位杂 交技术开始起步并得到运用且不断地发展起来。
荧光原位杂交

荧光原位杂交目录1荧光原位杂交2荧光原位杂交(FISH)技术详解3荧光原位杂交技术的发展历程1荧光原位杂交简介荧光原位杂交方法是一种物理图谱绘制方法,使用荧光素标记探针,以检测探针和分裂中期的染色体或分裂间期的染色质的杂交。
荧光原位杂交(fluorescence in situ hybridization, FISH)是在20世纪80年代末在放射性原位杂交技术的基础上发展起来的一种非放射性分子细胞遗传技术,以荧光标记取代同位素标记而形成的一种新的原位杂交方法,探针首先与某种介导分子(reporter molecule)结合,杂交后再通过免疫细胞化学过程连接上荧光染料.FISH的基本原理是将DNA(或RNA)探针用特殊的核苷酸分子标记,然后将探针直接杂交到染色体或DNA纤维切片上,再用与荧光素分子偶联的单克隆抗体与探针分子特异性结合来检测DNA序列在染色体或DNA纤维切片上的定性、定位、相对定量分析.FISH具有安全、快速、灵敏度高、探针能长期保存、能同时显示多种颜色等优点,不但能显示中期分裂相,还能显示于间期核.同时在荧光原位杂交基础上又发展了多彩色荧光原位杂交技术和染色质纤维荧光原位杂交技术.。
应用背景对于利用rRNA的荧光原位杂交来说,如下原因可导致较低的荧光信号强度:较低的细胞核糖体含量较低的细胞周边的通透性较低的目标序列可接触性(由于rRNA的折叠产生的构象,有些位置与rRNA分子内其他链或其他rRNA或蛋白紧密接触,从而使探针无法和目标序列杂交)为检验细胞中的目标序列是否容易被探针杂交,及测试最佳杂交温度,可利用“克隆荧光原位杂交”(clone-FISH)进行试验:将rRNA基因结合入质粒,转化至大肠杆菌中表达,构成核糖体,再用荧光标记的探针杂交。
FISH可与流式细胞术联用,对特定荧光标记的细胞进行计数或者分离。
[1]变体酶联荧光原位杂交(CARD-FISH)2荧光原位杂交(FISH)技术详解1974年Evans首次将染色体显带技术和染色体原位杂交联合应用,提高了定位的准确性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
荧光原位杂交-更新
荧光原位杂交(Fluorescence in situ hybridization,FISH)是一种基于DNA分子杂交技术,可以用于研究细胞核内DNA序列的空间分布,基因表达与功能等问题的一种重要方法。
相比于传统的DNA杂交方法,荧光原位杂交具有高灵敏度、高特异度、高分辨率、无需PCR扩增等优点,被广泛应用于在形态学及遗传水平对真核生物的相关研究。
本文将介绍荧光原位杂交的原理、方法和应用。
一、原理
荧光原位杂交的原理是利用标记有荧光染料(如荧光素、rhodamine等)的DNA探针与待检测样品(常为细胞核、染色体、tissue或者section等)中的目标DNA特异性结合,形
成稳定的探针-靶DNA杂交物,通过检测荧光发射信号的方法
来确定目标DNA序列的位置和数量。
探针的设计是荧光原位杂交成功的关键,因为它们必须
具有真确的互补性,绑定到目标DNA的特定区域上。
如何选择探针的特异性,通常取决于所要研究的问题,例如检测某一基因的副本数,探测非编码RNA,或发现肿瘤细胞中的染色体异
常等。
二、方法
1. 获取样品
荧光原位杂交技术所需的样品通常以细胞核或组织切片
的形式存在,依据所要研究的问题,通过相应方法处理,如:细胞核的分离、组织的固定剂的处理、剪切不同的组织块、制
备Paraffin等经典样品处理方法。
2. 样品前处理
对于切片和细胞各种杂质如化学物质、染料、蛋白质等的影响,靠的是样本的处理方法。
主要有以下几种:
1) 催化游离的核酸:
这一步的主要目的是去除待测样品中的核酸,包括double-stranded 的DNA和single-stranded的RNA。
当使用非常规杂交试剂或非常规探针(如bacterial artificial chromosome、cosmid probe)时,可能需要使用一些高效的去掉毛糙杂质的试剂。
2) 前处理(预处理):
固定、脱水、变性、去除RNA、去除单链DNA(ssDNA)、吉姆萨染色、荧光染色、脱色剂去除等多项操作。
3) 样品的杂交
根据探针的性质,杂交的方式也会有所不同。
若使用短的特异性DNA探针,则采用液态杂交技术。
液态的探头完全可溶于杂交缓冲液中,将探针和待测样品充分混合,使它们能够自由地通过半透膜进入细胞核。
当然,探针也可以使用固相杂交技术。
这种杂交的探针是直接黏附在玻片上的,待检测样品在特定条件下通过探针探测,降低了杂交的渐变,提高了杂交的稳定性和灵敏度。
三、应用
荧光原位杂交可以进行多种应用,可以用来检测基因或染色体缺失、异位、扫描等。
如果收集良好的样本,且如上所述的倒置显微镜和荧光标记是可用的,则下面是某些荧光原位杂交的典型应用。
1.检测某一基因的副本数
荧光原位杂交可用于检测一段DNA序列是否出现在某些
染色体上或基因组的其他区域。
其中一个典型的例子是检测HER2在乳腺癌中的拷贝数异常。
HER2 mRNA 受到扩增会导致HER2蛋白表达被扩大,从而加速乳腺癌的发展。
通过FISH可以检测出HER2基因在不同的细胞中是否扩增,进而帮助CT医生制定更好的治疗方案。
2.探测非编码RNA
FISH可以帮助研究者揭示RNA的分布、局部浓度和运动动力学行为,还可以测定RNA的空间组织以及一些细胞过程的可视化。
在某些细胞过程如细胞分裂、细胞衰老或炎症中非编码RNA(如lncRNAs、miRNAs等)发挥重要作用。
例如,在一项研究中,研究者使用荧光原位杂交方法鉴定了长链非编码RNA(LncRNAs,long non-coding RNAs)H19在基础表皮细胞癌中的表达模式。
3.研究肿瘤
荧光原位杂交技术在肿瘤研究中的广泛应用,因为它可
以准确地确定染色体的数量和染色体的缺失、附加或耗损,从而发现肿瘤的异常。
例如,在研究CLL白血病(CLL,chronic lymphocytic leukemia)时,FISH预测了这些患者有没有可
能具有13q-,12q-和del (11q)等染色体的异常。
四、总结
荧光原位杂交技术是一种强大而灵敏的检测技术,可用
于检测基因、核酸的空间分布、运动动力学和运动机制,以及一些细胞过程的可视化。
对于肿瘤抑制基因、肿瘤学、基因表达调控和生物学等领域的研究者而言,荧光原位杂交技术是一个不可或缺的工具。
近年来,随着生物学研究和临床医疗的需
求日益提高,荧光原位杂交技术的改良和运用也越来越广泛,必将有更多的创新和发展。