利用二代测序技术检出成人型多囊肾PKD1基因新突变
多囊肾的分子遗传、发病机制及治疗的研究进展1.

医学机能实验学综述论文题目:多囊肾的分子遗传、发病机制及治疗的研究进展班级:2010级口腔班姓名:闫子玉学号:2010508060262012年12 月 5 日多囊肾的分子遗传、发病机制及治疗的研究进展闫子玉综述(青岛大学医学院,2010级本科生)摘要:多囊肾是最常见的常染色体遗传病,有约50%最终发展为终末期肾功能衰竭。
近年来,该疾病的主要基因PKD1和PKD2的克隆测序陆续完成,对PKD的基因结构、分子发病机制以及治疗的研究取得了很大的确进展,本文主要对这些进展作以综述。
关键词:多囊肾病分子遗传学发病机制治疗研究进展引言:多囊肾病(polycystic kidney disease,PKD)是指双侧肾脏发生多个囊肿且进行性增大进而导致肾脏结构和功能损害的一种最常见的常染色体遗传性疾病。
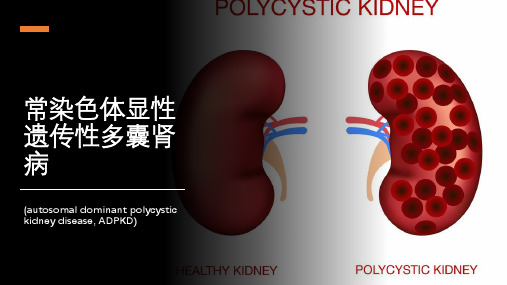
根据遗传方式不同, PKD可分为常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)和常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)。
ARPKD 多见于婴儿和儿童,发病率1/40000,多数早年夭折,很少存活至成年;ADPKD 多在成年后发病,发病率1/1000~1/400。
PKD有50%最终发展为终末期肾功能衰竭,约占终末期肾功能衰竭病因的10%,故近年来多囊肾病成为国际肾脏病领域研究的热点。
为早日攻克多囊肾病,国际上成立了多个协作组,如美国的多囊肾病研究基金会(PKR Foundation)[1]。
近年来也有许多研究表明该病可能为一种在易感人群中发生的感染性疾病。
随着分子生物学的发展,人类对多囊肾病有了一定的认识。
现综述如下。
一.先天性多囊肾分子遗传学基础目前已知的有3种基因突变导致了常染色体显性遗传多囊肾病,据报道其中2种最为常见,PKD1所占比例约85%;PKD2所占比例约15%;还有一种是PKD3,所占比率很小[2] 。
《多囊肾病》PPT课件

两项主要+一项次要
第一项主要+三项以上次要
医学PPT
17
诊断
3、影像学检查: Ravine等于1994年提出了一下B超诊断标准:
有家族遗传史的30岁以下患者,单侧或双侧肾脏 有2个囊肿,30~59岁患者双侧肾脏至少各4个囊 肿;如果同时伴有其他肾外表现,如肝囊肿等, 诊断标准可适当放宽。此诊断标准敏感性97%, 特异性90%。如无家族遗传史,每侧肾脏有10个 以上囊肿,并排除其他肾囊肿性疾病方可诊断
多囊肾病
(polycystic kidney disease,PKD)
医学PPT
1
• 肾囊肿:包曼囊~集合管间的局部扩张→与 原来的肾小管脱离→独立、充满液体的囊肿
• 囊性肾脏病:肾脏出现单个或多个囊肿的一 组疾病,分遗传性和非遗传性
医学PPT
2
医学PPT
3
多囊肾的分型
目前已知有3种基因的突变导致 了ADPKD
其中2种最为常见: 为PKD1 (约 占85% )和PKD2 (约占15% )
第3种是PKD3
医学PPT
4
多囊肾的分型
• 常染色体显性遗传型,( ADPKD)此型一般到成年 才出现症状,又称成人型 多囊肾,实际上可发生于 任何年龄,故“成人型”并 不准确,已废用
• 常染色体隐性遗传型,又 称婴儿型多囊肾. 一般在 婴儿即表现明显
医学PPT
13
发病机制2、螺旋区-螺旋区相互作 用假说
医学PPT
14
发病机制3、纤毛致病学说(中心地位)
纤毛无运动功能,必须依赖一个非常重要 的生理过程———鞭毛内运输(IFT) 来完成, 驱动蛋白- 和细胞浆动力蛋白- 1B 作为2种 分子动力蛋白, 分别参与其中的顺向转运和 逆向转运过程,纤毛通过多囊蛋白复合体感 受尿流率调控肾小管腔直径和分化状态, 基 因突变引发了纤毛感受尿流率的功能异常, 可通过下游Ca2 +信号传导影响靶基因调控 ,进而导致细胞功能改变和囊肿生成
关于胡桃夹综合征与多囊肾

胡桃夹综合征(nutcracker phenomenon)即(左肾静脉压迫综合征),又称胡桃夹现象,好发于青春期至40岁左右的男性,儿童发病分布在4~7岁,多发年龄见于13~16岁。
人体的血管像四通八达的道路一样,是有一定走向的。
左肾静脉行走在腹主动脉和肠系膜上动脉之间,这两条动脉构成40~60度的夹角,左肾静脉刚好通过此夹角。
从解剖上看,右肾静脉径直注入下腔静脉,行程短而直。
而左肾静脉则需穿过腹主动脉和肠系膜上动脉之间的夹角,跨越腹主动脉前方始能注入下腔静脉,因此左肾静脉远较右肾静脉长。
正常时,肠系膜上动脉与腹主动脉之间的夹角被肠系膜、脂肪、淋巴结和腹膜等所充塞,使左肾静脉不致受到压挤。
当青春期发育较快、身高迅速增长、脊柱过度伸展、体形急剧变化或肾下垂等情况下,左肾静脉在这个夹角中的日子就不好过了,会受到挤压,引起血流变化和相应的临床症状。
症状:胡桃夹现象的主要症状是血尿和蛋白尿,其中无症状肉眼血尿更易发现。
血尿的原因是左肾静脉受压致肾静脉高压,左肾静脉扩张所引流的输尿管周围静脉与生殖静脉淤血、与肾集合系统发生异常交通,或部分静脉壁变薄破裂,引起非肾小球性血尿,还会发生睾丸静脉和卵巢静脉淤血而出现肋腹痛,并于立位或行走时加重。
另外男性还能发生精索静脉曲张。
此外有蛋白尿,不规则月经出血,高血压等。
此病的诊断标准为:一侧肾出血;尿红细胞形态为非肾小球性;尿中钙排泄量正常;膀胱镜检查为左侧输尿管口喷血或血性尿;腹部彩超或CT检查可见左肾静脉扩张等。
超声检查:超声对胡桃夹综合征的诊断有着明显的优势,超声检查时可清晰显示腹主动脉、肠系膜上动脉及左肾静脉的解剖情况,在不同横断面均可找到左肾静脉扩张近段的最大内径,测值准确,同时可观察并测量肠系膜上动脉与腹主动脉夹角变化。
彩超血流速度提供更准确的血流动力学变化,有助于此病诊断。
超声检查还能除外先天性畸形、外伤、肿瘤、结石、感染性疾病及血管异常等造成的血尿。
多囊肾多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。
常染色体显性遗传性多囊肾病
流行病学ห้องสมุดไป่ตู้
•
所有种族都可发生ADPKD,报道的患病率为1:1000。在美国每年
开始透析的患者中,大约有5%的肾病由ADPKD引起。
•
ADPKD主要由两种基因突变引起:16号染色体上的PKD1(编码多
囊蛋白-1)或4号染色体上的PKD2(编码多囊蛋白-2)。大多数患者可维持
正常肾功能到30岁左右。一旦GFR开始下降,其每年可平均降低4.4-
➢ 多囊肝 多数不需治疗。肝脏明显增大可引起腹胀、呼吸困难、胃食管反流、门静脉高压等。可根据病情选 择肝囊肿穿刺硬化、去顶减压术、肝部分切除术或肝移植术
➢ 颅内动脉瘤 对于有动脉瘤和蛛网膜下腔出血家族史的病人,推荐MRI血管造影检查确诊。直径大于10mm的动 脉瘤应采取介入或手术治疗
治疗
肾脏替代治疗 ➢ 包括血液透析、腹膜透析和肾移植。目前认为ADPKD病人腹膜透析与血液透析的并发症和 长期存活率无明显差异。移植后肾存活率、并发症与其他肾移植人群相似
治疗
对症治疗 ➢ 疼痛
急性疼痛针对病因进行治疗。慢性疼痛,程度轻者或一过性疼痛卧床休息并观察,如疼痛持续或较 重按止痛阶梯序贯药物治疗,仍不能缓解可考虑囊肿穿刺硬化、囊肿去顶减压术及多囊肾切除术
➢ 出血 多为囊肿出血所致,呈自限性,轻者绝对卧床休息、止痛、多饮水。出血量大、保守疗法效果差可 行选择性血管栓塞或出血侧肾脏切除
泌尿道和囊肿感染,引起ADPKD病人发热的首要病因,主要表现为膀胱炎、肾盂肾炎、囊肿感 染和肾周脓肿,逆行性感染为主要途径
临床表现
肾外表现 ➢ 囊性病变
可累及肝脏、胰腺、脾脏、卵巢及蛛网膜等器官。其中肝囊肿最常见,大多数病人无症状,少数 可表现为疼痛、囊肿感染和出血
多囊肾

特殊治疗
• 1、免疫诊断 • 2、免疫清除 • 当肾功能进展至尿毒症期,可以应用免疫
清除疗法,在透析的基础之上,应用透析 滤过,血液滤过,组合型人工肾及床旁血 滤等,更好地清除除中小分子之外的中大 分子毒素及细胞因子、炎症介质等,更好 地控制并发症的出现,提高患者的生活质 量。
特殊治疗
• 3、免疫阻断 • 如果患者合并肾小球肾炎,有时会出现大
辅助检查
• .病理改变 显微镜观察可见正常肾组织受到邻近 囊肿的压迫,在血管硬化或肾盂肾炎的基础上继 发肾小球硬化、小管萎缩和间质纤维化。对囊肿 的组织来源的鉴定较困难,除非囊肿保持了原组 织的正常位置和上皮的形态学特点。来自肾小囊 的囊肿有时含有变形的小襻状肾小球血管丛;源 自深部集合系统的囊肿常为薄壁;发生于包膜下 集合系统的囊肿壁较厚,常包绕有致密纤维结缔 组织。可用特异植物凝集素结合试验帮助鉴定囊 肿的组织来源是近端小管、集合管或其他。
辅助检查
• 血常规、尿常规、便常规、肝功、肾功、 血糖、血脂、电解质、C反应蛋白、凝血五 项、内生肌酐清除率、心电图、胸部正位 片、肝脏肾脏彩超
• 肾损十项、肾脏CT、肾脏ECT、淋巴细胞 亚群、免疫五项、自身抗体系列
诊断及鉴别诊断
• 在症状轻的患者,本病常误诊为单纯性肾囊肿、 孤立性多房囊肿等多发性单纯囊肿,家族史和同 时存在的肝囊肿可帮助鉴别诊断。发生血尿者须 与新生物、肾结石等引起血尿的其他疾病进行鉴 别,注意多囊肾并发结石或囊肿癌变等情况,应 行凝血筛查(PT、APTT 和血小板),排除出血性 疾病。对于有蛛网膜下腔出血家族史的患者可行 脑血管MRI 检查。
一般治疗
• 4.防治尿路感染及肾结石 选择有效抗生素积极给 予抗感染治疗,主要目的是用于治疗并发的急性 肾盂肾炎,少数出于预防性用药。本病患者易发 生尿路感染,尤其是女性,如诱发肾盂肾炎或囊 肿感染则肾区疼痛加重伴发热,血尿及脓尿明显 ,严重者可导致败血症。因此,必须积极治疗。 抗菌药物应选用易进入囊肿腔内的药物,磺胺甲 噁唑/甲氧苄啶(SMZ-TMP)及喹诺酮类,均易进 入近端及远端小管的囊肿腔内。如系近端小管的 囊肿常选用青霉素类和头孢菌素类。多饮水、勤 排尿、尿液碱化是预防结石及钙化的重要措施。
肾囊性病变CT诊断

21
肾结核 (renal tuberculosis)
肾结核早期在乳头部或髓质锥体的深部形成结核结节 或肉芽肿,中心发生干酪坏死,随着病变扩展与肾盏相通, 坏死物质经肾盏排出,形成空洞,病变进一步扩展可自一 个肾盏至一组肾盏,成为肾盂结核。
根据病情的发展不一样,形态学可分为:结节型,囊 肿型,积水型,积脓型,萎缩型,钙化型,混合型。
增强扫描可表现为边缘环形强化并内部分隔 明显强化。静止期或寄生虫死亡时可出现钙化。 治疗及预后:
手术治疗是最有效的治疗方法。
精选ppt课件
29
(a) 平扫右肾复杂性囊性肿块 (b) 增强CT扫描病变环形强化伴分隔明显强化
精选ppt课件
30
精选ppt课件
31
肾小球囊肿病 (glomerulocystic kidney disease, GCKD)
精选ppt课件
19
精选ppt课件
20
肾脓肿 ( renal abscesses )
肾实质感染所致广泛的化脓性病变,或输尿管梗阻后 肾盂肾盏积水、感染而形成的一个集聚脓液的囊腔。
早期肾实质内略低密度影,轻度不 规则强化。
成熟期为类圆形均匀低密度影,增 强呈低密度影周围伴环形强化。
成熟期脓肿
精选ppt课件
男性单侧发病较多;女性则以双侧发病较多,且往往 左肾受累更明显。
精选ppt课件
38
患肾呈不规则分 叶的多发囊肿或 呈葡萄状,几乎 看不到肾实质。
精选ppt课件
39
肾脏多发葡萄串样囊变,囊肿间有互不相通的分隔, 分隔或少量实体部分可强化。
精选ppt课件
40
获得性肾囊性疾病 (acquired cystic kidney disease,ACKD)
遗传性肾病的基因检测及其治疗技术研究

遗传性肾病的基因检测及其治疗技术研究随着人类基因科技的迅猛发展,遗传性疾病的基因检测和治疗技术也日益成熟。
其中,遗传性肾病是一种常见的家族性疾病,对患者及家庭造成了巨大的负担,因此对其的基因检测及治疗技术研究显得尤为重要。
一、遗传性肾病的类型遗传性肾病按照基因突变的部位和影响,可以分为单基因遗传和多基因遗传两类。
单基因遗传包括最为常见的ADPKD(成人多囊肾病)、ARPKD(儿童多囊肾病)、Alport综合征等;而多基因遗传包括肾小管间质病、先天性肾小球性疾病等多种。
二、基因检测技术随着基因检测技术的逐渐成熟,已经可以通过对患者家族史及对基因的检测,来判断患者是否患有遗传性肾病。
其中,ADPKD 患者中,大约85%有PKD1基因的突变。
因此,对于成年的ADPKD患者,仅需检测PKD1基因,即可对其进行准确的判断;而对于18岁以下的患者,还需检测PKD2基因。
ARPKD患者则需要检测PKHD1基因的突变。
基因检测是一种无创、高效的诊断方法,可以让患者尽早接受到合适的治疗。
三、遗传性肾病的治疗技术目前,遗传性肾病的治疗技术主要包括保守治疗、手术治疗和药物治疗三种。
1. 保守治疗保守治疗主要是通过控制高血压、糖尿病等基础疾病,使肾脏不受进一步伤害。
但对于多数遗传性肾病患者而言,如果不采取其他治疗措施,肾脏功能会逐渐下降,甚至最终导致肾衰竭。
2. 手术治疗手术治疗主要包括硬膜外脑室分流术、肾移植等。
硬膜外脑室分流术是一种可以减轻颅内压力、改善脑功能的手术,适用于脑积水等疾病。
而肾移植则是目前治疗肾衰竭的常规治疗方法,对于遗传性肾病患者,肾移植可以起到延缓肾脏损害的作用。
3. 药物治疗药物治疗主要是通过调节肾脏的生理功能,延缓肾脏的进一步损害。
而目前,新一代的基因治疗技术也已经开始应用于临床,对于ADPKD等囊肿性肾病患者,可以通过小分子化合物等药物来针对性地抑制囊肿增长。
四、结语遗传性肾病是一种严重的疾病,对患者、家庭和社会造成了极大的负担。
(医学课件)多囊肾演示课件

中医药治疗多囊肾的研究及进展
中药方剂
采用中药方剂,如补肾活血汤等,调节患者免疫功能,改善肾功能。
针灸理疗
结合针灸、推拿等理疗方法,缓解症状,提高患者生活质量。
05
预后及预防
成人型多囊肾的预后情况
预后较好
在青年时期,患者往往没有明显的症状,肾功能也正常,预后 较好。
肾功能减退
随着年龄的增长,囊肿会逐渐增大,肾功能会逐渐减退,出现 高血压、尿毒症等严重并发症。
01
基因突变致多囊肾疾Βιβλιοθήκη 遗传方式多种多样,包括常染色体显性
遗传、常染色体隐性遗传、X连锁隐性遗传等。
肾功能受损
02
患者肾功能受损程度差异较大,有的患者肾功能正常,有的则
出现严重肾功能不全。
其他症状
03
患者还可能出现眼部异常、颅面部畸形、心血管疾病、神经系
统疾病等多种症状。
03
诊断与鉴别诊断
成人型多囊肾的诊断标准
单纯肾囊肿通常无症状,囊肿体积 较小,无家族遗传史。
与肾盂肾炎的鉴别
肾盂肾炎常伴有发热、尿路感染等 症状,尿液检查可发现尿路感染的 证据。
与肾脏肿瘤的鉴别
肾脏肿瘤通常为实质性肿块,生长 迅速,可伴有疼痛、血尿等症状。
与慢性肾衰竭的鉴别
慢性肾衰竭常伴有高血压、贫血、 电解质紊乱等全身症状,肾功能检 查异常。
支持治疗
对于已经确诊的多囊肾患者,应该积极控制 血压、血糖等并发症,延缓病情进展。
对于晚期多囊肾患者,应该采取支持治疗措 施,如透析、药物治疗等,以缓解症状和提 高生活质量。
06
研究进展及展望
多囊肾的基因治疗研究进展
01
基因治疗为多囊肾的治疗开启了新的方向,特别是对于无法进行手术或药物治 疗的患者。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
术通讯作者:宋畴(Email.'songf-558固263.net) 背景Angelman综合征(Angelman syndrome,AS)和Pralli
syndrome,PWS)分别是由于15q11.2-q13区内母源或父源等位
基因印记异常所致的多系统复杂性遗传病,两者发病率均约为1/1,5000。两种 疾病的遗传机制较类似,而临床特点各不相同。15q11.2-q13区的缺失、单亲二 倍体(uniparental disomy,UPD)以及印记缺陷是两者的遗传学发病机制,区 别仅在于发生在父源或母源区域。另外,AS还可由位于母源15q11.2-q13区的
(Nimblegen 2.1 M)进行杂交,富集PKDl基因的全部编码区外显子及其邻接内
含子区域(外显子旁侧100bp),最后利用HiSeq2000(Illumina,San Diego,USA) 测序系统进行测序, 对测序读取的90 bp短序列使用BWA(Burrows Wheeler Aligner)软件包将过滤后的读序比对至NCBI数据库(Build 37)参考序列上,分 别使用SOAPsnp矛NGATK Indel软件分别对单核苷酸多态性(SNP)位点和小的插 入和缺失(Indel)进行数据分析。我们在一个女性患者PKDl基因的外显子15
2.35
Angelman/Prader—Willi综合征的基因诊断和临床分析
首都儿科研究所遗传研究室(100020)白晋丽瞿宇晋宋叻 首都儿科研究所附属儿童医院(100020)王立文陈晓波 解放军总医院儿科中心(100853)邹丽萍 北京大学第一医院儿科(100034)杨艳玲 首都医科大学附属北京儿童医院神经科(100045)杨欣英
204
区域的补充,实现对PKl)I的全部编码外显子区域全覆盖测序分析,简化了PKDl
基因的基因诊断程序。另外,通过存储的频率信息可以进一步评估其致病可能, 节省了后期对于新发突变在群体发生频率的验证工作。在患者中检出一致病突 变,可作为其生育时产前基因诊断的依据。本研究检出的新的突变类型补充了 PKDl基因突变数据库。
泛素连接酶3A基因(删)突变直接所致。目的通过对临床诊断或疑似的
AS/PWS患儿进行遗传学分析,使患儿获得基因诊断、了解两种疾病的遗传缺陷 类型分布以及临床特点。方法(1)按照AS和PWS临床诊断评分标准入选临床诊断 或疑似的91例AS患儿和60例PWS患儿。(2)采用甲基化特异性PCR(MS—PCR)、 短串联重复序列(STR)连锁分析、LIBE3A基因序列分析(仅针对As疑似病例) 技术和染色体核型分析同时进行AS和PWS的基因诊断和遗传机制分析。(3)在遗 传学检测的同时,进行相关内容的问卷调查。结果(1)所有As和PWS病例均为散 发病例,父母非近亲婚配。(2)48例患儿明确诊断为AS,占入组病例的52.7% (48/91)。其中40例为15q11.2—13区域的母源缺失(83.3%,40/48),2例为 父源单亲二倍体型(4.2%),2例为印记缺损型(4.2%),2例发生了L猩E3A基因 突变(4.2%):Pr0400HiS和Asp563Gly。有2例患儿父母未提供DNA样品,未进 行遗传机制分型。对资料完善的35例AS阳性患儿进行临床分析,结果显示男性、 女性患儿分别为16例和19例,年龄范围8月"--'6岁3月,平均诊断年龄27.8 月。患儿平均竖头、独坐和独走年龄分别为5.5、11.6和33.8月。出生时身长、 体重及头围均在正常参考值范围。就诊时,所有的患儿均可观察到精神运动发育 迟缓、语言障碍、运动或平衡障碍和快乐表情等表现,近90%患儿出现癫痫和特 征性脑电图(典型棘波、慢波),仅约30%患儿存在小头畸形,20%--80%患儿存 在AS的相关性表现或体征,尤以平枕或枕骨凹陷、宽嘴,宽牙缝等较突出。(3) 获得基因诊断的PWS阳性患儿共22例,占入组病例的36.7%(22/60):15例为 15q11.2—13区域的母源缺失(68.2%,15/22),5例为父源单亲二倍体型(22.7%),
2.34
利用二代测序技术检出成人型多囊肾PI(D1基因新突变
杨涛1,孟岩1,张明荣2,王炜辰1,魏晓明2,黄尚志1
2.
中国医学科学院基础医学研究所,北京协和医学院基础学院 3.深圳华大基因研究院
常染色体显性遗传性多囊肾病f
autosomal dominant polycystic kidney disease.
205
ADPKD)是最常见的遗传性肾脏病,其发病率为1/400.1/1000,在终末期肾功能 衰竭患者中占5-8%。该病以双侧肾脏发生多个囊肿且进行性增大为特征,约50% 的患者最终可发展为肾功能衰竭。其遗传病因源于PKDl或PKD2基因的突变。 通常由PKDl基因突变所致的ADPKD患者的临床症状较重,发生肾功能衰竭的 年龄较早。在ADPKD家系中,PKDl突变占85%,PKD2突变占15%。PKDl 基因包含12906bp的编码区,含46个外显子。以往多采用逐个外显子区域PCR 扩增Sanger测序的方法,非常繁琐。另一方面,上游的33个外显子区域在16 号染色体上有多个同源重复,部分区域的重复次数可高达14次以上,这也给 PKDl基因的突变检测造成很大的难度。因此,建立更为高效准确的PKDl基因 突变检测方法尤为重要。近年来出现的目标基因捕获和新一代测序技术有可能为 PKDl基因突变检测带来技术上的突破。 我们和深圳华大基因研究院合作,利用该项技术对在PKD2基因中未检测出 致病突变的两个ADPKD患者进行了PKDl基因测序研究。血液中提取的基因组 DNA经CovariS S2超声打断仪将DNA打断至200、300bp后,连接特异性标签序列, 按照1notyping v.2,
http://genetics.bwh.harvard.edu/pph2)对P.Glyl3 19Arg致病性进行预测,结果均判 定为有害突变。以上证据均支持P.Glyl319Arg为致病突变。在另一位患者的外显 子15区域检出一杂合碱基改变c.5847C>T,为1949位Ser同义突变 (p.Serl949Ser),文献已报道为无致病性的SNP(rs801 1 1665),二代测序数据统计 学分析也排除了其致病性。为了避免可能出现的漏检,我们对测序深度<8的外 显子1、2和3区域建立Sanger测序的方法,仍然没有检出突变。 我们通过目标区域捕获二代测序技术并结合Sanger测序对少数读序低深度
区域检出一杂合碱基改变c.3955G>A,并用Sange湖0序验证了该突变。患者父母
表型正常,无相同碱基改变,患者为新生突变。由于本突变未见文献报道,我们 进一步对突变的致病性进行了分析。c.3955G>A造成蛋白质编码的1319位中性 的甘氨酸变成了碱性的精氨酸(p.Glyl319Arg),有可能较大地影响蛋白质的空间 结构;1319位于重要的PKD结构域,是蛋白质互作过程中可能的配体结合位点; 对455位正常无关个体相应区域测序未检出同样SNP;在10种哺乳动物中进行 了蛋白质保守性分析显示p.Glyl319位点高度保守。经SIFT在线预测工具 (http://sift.jcvi.org/)以及PolyPhen一2软件fPolymorphism