紫外可见吸收光谱计算
紫外吸收和漫反射光谱转换

紫外吸收和漫反射光谱转换
紫外吸收和漫反射光谱转换是通过特定的数学公式,将测得的漫反射光谱数据转换为吸收光谱数据的过程。
紫外可见吸收/漫反射光谱(UV-Vis Spectroscopy)是一种分析技术,它利用物质对光的选择性吸收或反射特性来测定物质的组成、含量及结构。
在实际操作中,尤其是对于固体样品,通常会测量其漫反射光谱,然后使用Kubelka-Munk公式将这些数据转换为吸收光谱。
具体步骤如下:
1. 测量漫反射光谱:首先,使用紫外可见漫反射光谱仪测量样品的漫反射光谱。
这涉及到照射样品并测量反射光的强度。
2. 应用Kubelka-Munk公式:然后,使用Kubelka-Munk公式将漫反射光谱数据转换为吸收光谱数据。
这个公式是基于漫反射光谱的理论推导出来的,它可以将反射率转换为与吸收系数相关的值。
3. 计算吸收光谱:最后,根据Kubelka-Munk函数,可以通过漫反射光谱计算出吸收光谱,从而得到样品的吸收特性。
需要注意的是,这个过程需要一定的数学和物理知识,以及对相关仪器的操作熟悉度。
在实际操作中,可能还需要考虑样品的特性、测试条件等因素,以确保转换的准确性和可靠性。
紫外和可见光吸收光谱

紫外和可见光吸收光谱1.紫外光谱及其产生⑴紫外光的波长范围紫外光的波长范围为4-400nm。
200-400为近紫外区,4-200nm为远紫外区。
由于波长很短的紫外光会被空气中氧和二氧化碳吸收,研究远紫外区的吸收光谱很困难,一般的紫外光谱仅仅是用来研究近紫外区的吸收。
⑵紫外光谱当把一束光通过有机化合物时,某一波长的光可能吸收很强,而对其他波长的光可能吸收很弱,或者根本不吸收。
当化合物吸收一定波长的紫外光时,电子发生跃迁,所产生的吸收光谱叫做紫外吸收光谱,简称紫外光谱。
⑶电子跃迁的种类在有机化合物分子中,由于化合物的价电子有三种类型,即σ键电子、π键电子和未成键的 n 电子,在电子吸收光谱中,电子跃迁主要是经下三种。
①σ-σ*跃迁σ电子是结合得最牢固的价电子,在基态下,电子在成键轨道中,能级最低,而σ*态是最高能级。
σ-σ*跃迁需要相当高的辐射能量。
在一般情况下,仅在200nm以下约~150nm才能观察到,即在一般紫外光谱仪工作范围之外,只能用真空紫外光谱仪才可观察出来(在无氧和二氧化碳的情况下)。
所以测紫外光谱时,常常用烷烃作溶剂。
② n电子的跃迁n 电子是指象N,S,O,X 等原子上未共用的电子。
它的跃迁有两种方式。
第一种方式:n-π* 跃迁未共用电子激发跃入π*轨道,产生吸收带,称为R带(基团型的,Radikalartig德文),由n-π*引起的,在200 nm以上。
如:醛酮分子中羰基在275-295nm处有吸收带,为C=O中n-π*跃迁吸收带。
第二种方式是n→σ*跃迁,这种跃迁所需的能量大于n-π*,故醇醚均在远紫外区才出现吸收带。
~ 200nm。
如甲醇λmax183nm。
③π→π*跃迁乙烯分子中π电子吸收光能量,跃迁到π*轨道。
吸收带在远紫外区。
当双键上氢逐个被烯基取代后,由于共轭作用,π→π*能级减小。
吸收带向长波递增。
由共轭双键产生的吸收带称为K带,其特征是摩尔消光系数大于104。
在近紫外区吸收,CH2=CH2 λmax162nm,CH2=CH-CH=CH2 λmax217nm。
第九章 紫外吸收光谱分析
3.在下列化合物中,哪些适宜作为紫外 光谱测定中的溶剂? 甲醇、乙醚、苯、碘乙烷、乙醇、 正丁醚、环己烷 4. 下列化合物中哪一个的max最长? CH4; CH3I; CH2I2
在下列化合物中同时含有*、 n*、 *跃迁的化合物是 三氯甲烷、丙酮、丁二烯、二甲苯
在下列化合物中,那一个化合物能吸 收波长较长的辐射( ) 苯、二甲苯、对氯代甲苯、萘
1, 3-丁二烯:max=210nm, =20000L· mol-1· cm-1
1, 5己二烯:两个不共轭的双键,1-己烯:一个双键。 1, 5-己二烯与1, 3-丁二烯比较:两者都有两个双键, 摩尔吸光系数相近;区别: 1, 3-丁二烯中两个双键共
轭,吸收波长红移,最大吸收波长= 210nm 。因此,
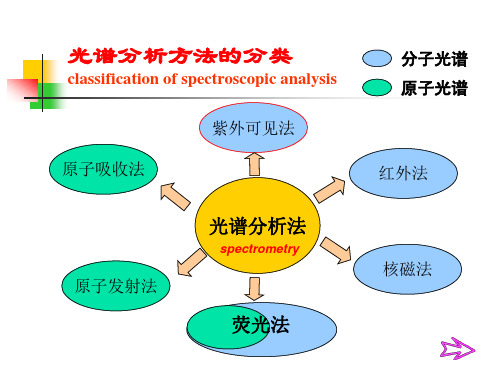
光谱分析方法的分类
classification of spectroscopic analysis 紫外可见法
分子光谱 原子光谱
原子吸收法
红外法
光谱分析法
spectrometry
原子发射法
核磁法
荧光法
光学分析法概要(P201)
依据:物质吸收、发射电磁辐射(电磁波;光) 光学分析法:利用物质与电磁辐射的相互作用来进行 分析的方法。
⑶ * 跃迁(NV跃迁)
吸收波长处于远紫外区的近紫外端或近 紫外区,max一般在104以上, 强吸收。有机化 合物中含有 电子的化合物均可发生该类跃 迁。如不饱和烃 * 跃迁 ( 乙烯 * 跃 迁的max=165nm, max=104;乙炔*跃迁的 max=173nm 。 乙 醛 * 跃 迁 的 max 为 190nm,max:104。( <200nm ;生色团)
某化合物分子式为,
紫外吸收光谱

R带与K带
R带: R带相当于n →π* 跃迁所吸收的能量产生的吸收带。 含有杂原子的不饱和基团,如-C=O、-N=N- 、-NO、-NO2 等发色团的特征。 特点:(1)吸收较弱; (2)波长较长270-300nm K带: 由于共轭双键中π → π* 跃迁所产生的吸收带称为K带。 特点:(1)吸收强度大,摩尔吸光系数大(104-105之间) (2)波长在217-280nm之间。 (3)利用紫外吸收光谱是否有K吸收带,作为判断共 轭体系的重要依据。
讨论:
(4)吸收光谱波长分布由产生谱带跃迁能级间的能量差决定, 反映分子内部能级分布状况,是物质定性依据; (5)吸收谱带强度与分子偶极矩变化、跃迁几率有关,提供分 子结构信息。通常将在最大吸收波长处测得的摩尔吸光系数 ε max作定性依据。不同物质λ max可能相同,但ε max不一定
相同;
(6)吸收谱带强度与该物质分子吸收光子数成正比,定量分析
紫外吸收光谱
电子跃迁与分子吸收光谱
物质分子内部三种运动形式: (1)电子相对于原子核的运动; (2)原子核在其平衡位置附近的相对振动;
(3)分子本身绕其重心的转动。
分子具有三种不同能级:电子能级、振动能级和转动能级 三种能级都是量子化的,且各自具有相应的能量。 分子的内能:电子能量Ee 、振动能量Ev 、转动能量Er 即: E=Ee+Ev+Er
生色团 烯 炔 羧基 酰胺基 羰基 偶氮基 硝基 亚硝基 硝酸酯
溶剂 正庚烷 正庚烷 乙醇 水 正己烷 乙醇 异辛酯 乙醚
二氧杂环己烷
/nm 177 178 204 214 186 339,665 280 300,665 270
emax
紫外最大吸收波长的计算方法

紫外最大吸收波长的计算方法紫外最大吸收波长的计算方法___________________________紫外(Ultraviolet)光的特性是与可见光不同的,它的波长比可见光更短,能够激发物质的电子进行激发态,因此有着重要的作用。
紫外光吸收谱中最大吸收波长是描述该物质对紫外光的吸收能力的重要参数,它主要取决于物质的分子结构,分子团及其环境。
本文主要介绍紫外最大吸收波长的计算方法。
一、紫外最大吸收波长的原理--------------------------------紫外最大吸收波长的计算主要是基于光谱学原理。
物质的分子具有一定的电子结构,当入射的光照射到物质分子时,分子中的电子会受到入射光的激发,由低能态跃迁到高能态,从而使物质分子发生变化,从而使物质产生吸收光谱。
其中,最大吸收波长表明该物质对紫外光的最强吸收能力。
二、紫外最大吸收波长的计算方法---------------------------------1. 通过仪器测量法来计算仪器测量法是一种常用的方法,它能够直接测量出物质对紫外光的最大吸收波长。
常用仪器如分光光度计、吸收光度计、旋光仪、衍射仪、偏振仪等,通过调整入射光波长,在发射或吸收光强度上变化的斜率可以计算出物质的最大吸收波长。
2. 通过理论计算方法来计算理论计算方法是通过物质的分子结构、电子能量层次、电子分子态、电子-电子相互作用和其它因素来对物质的吸收光谱进行理论模拟,从而估算出物质的最大吸收波长。
理论计算方法不仅能够准确地估算出物质的最大吸收波长,而且还可以准确地得到物质的其它吸收光谱特性,如共振强度、共振宽度、吸收强度和其它信息。
三、紫外最大吸收波长的应用---------------------------紫外最大吸收波长对于很多领域都具有重要的意义,如化学、材料、生物学、生态学、医学、农学、气候学等都有广泛的应用。
在化学方面,它可以帮助我们识别物质分子中包含哪些元素;在材料方面,它可以帮助我们识别材料中是否存在有害物质;在生物学方面,它可以帮助我们识别生物体中存在哪些物质;在医学方面,它可以帮助我们识别人体中是否存在某些有害物质。
紫外吸收光谱

化合物
1,3-丁二烯 1,3,5-己二烯
溶剂
己烷 异辛烷
max/nm
217 268 304 334 364
emax
21,000 43,000 — 121,000 138,000
1,3,5,7-辛四烯 环己烷
1,3,5,7,9-癸四烯 1,3,5,7,9,11- 十 二 烷基六烯
用不同波长的单色光照射, 测吸光度;
吸收曲线的讨论:
①同一种物质对不同波长光的 吸光度不同。吸光度最大处对应 的波长称为最大吸收波长λ max ②不同浓度的同一种物质,其吸收曲线形状相似λmax不变。 而对于不同物质,它们的吸收曲线形状和λmax则不同。 ③吸收曲线可以提供物质的结构信息,并作为物质定性分析的 依据之一。
,但ε max不一定相同;
(6)吸收谱带强度与该物质分子吸收的光子数成正比,定量分
析的依据。
一、紫外吸收光谱的产生
formation of UV
1.概述
紫外吸收光谱:分子中价电子能级跃迁。 波长范围:100-800 nm. (1) 远紫外光区: 100-200nm
(2) 近紫外光区: 200-400nm
讨论:
④不同浓度的同一种物质,在某一定波长下吸光度 A 有差异,在λ max处吸光度A 的差异最大。此特性可作作
为物质定量分析的依据。
⑤在λ max处吸光度随浓度变化的幅度最大,所以测定
最灵敏。
基本术语
生色团: 分子中可吸收光子产生电子跃迁的基团 最有用的紫外—可见光谱是由π→π*和n→π*跃迁 产生的。这两种跃迁均要求有机物分子中含有不饱 和基团。简单的生色团由双键或叁键体系组成,如 乙烯基、羰基、亚硝基、偶氮基—N=N—、乙炔基 、腈基—C≡N等。
紫外-可见光谱
取代苯 烷基取代苯:影响小,由于超共轭效应,导致红移,
降低B- 带的精细结构; 助色团取代苯:n 电子与苯环形成 p-π共轭,导致红移,
增强B-带的强度,降低B- 带的精细结构
连有推电子基团的红移强弱顺序为: CH3 < Cl < Br < OH < OCH3 < NH2 < O-
同环双键母体 253 两个延长双键 60 三个环外双键 15 五个取代烃基 25 —————————
353nm
IV
具有四个以上双键的共轭体系,K- 带λmax 和εmax 值按规则计算:
λmax = 114 + 5M + n(48.0-1.7n) – 16.5 Rendo -10 Rexo εmax = (1.74 x 104) n
红移 紫移
取代苯的λmax 值经验计算参数 λmax = 203.5 + 取代基位移值 (误差有时较大)
Scott 规则
多核芳香族化合物:总体红移。 多联苯以及苯并多环
杂环芳香族化合物
六元环:较苯的吸收加强;并产生 n→π* 跃迁;
五元环:类似双烯。
6. 影响紫外光谱的因素: 外部因素:
溶剂:纯度极其重要;极性影响复杂; 浓度:控制吸光度为0.7-1.2范围; 样品池:应不影响样品对紫外的吸收;
内部因素: 位阻:
平衡体系的紫外光谱:
一些极性化合物,在极性或pH 不同的溶剂中 光谱有较大的变化,如互变异构平衡及酸碱平衡:
中性
碱性
7. 紫外光谱解析
缺点:只提供分子中共轭体系和一些基团的结构信 息,不能推知分子结构。
有机化合物的紫外吸收光谱
R2 -C6H4 -COR
R1为烷基时的基本值 R1为H时的基本值 R1为OH时的基本值 R2为下列基团时
烷基
-OH -OR
-O-
-Cl
-Br
-NH2 -NHAc
-NR2
K吸收带波长λ/nm
246 250
230 邻位 间位 对位
3
3 10
7
7
25
11
20 78
0
0
10
2
2
15
13 13 58
20 20 45 20 20 85
εmax=1.74N× 104
式 中 A— 取 代 基 数 目
N— 共 轭 双 键 数
R—末端含双键的环数 E—环外双键数
三、芳族化合物的紫外吸收光谱
苯是最简单的芳香族化合物,它的紫外吸收光谱有三 个吸收带,其吸收波长分别为184nm(E1带 ε=47000)、 203nm(E2或K带ε=7000 )和256nm(B带ε≈200))。 B带的吸收强度比较弱,在非极性溶剂中或呈气体状态时出 现精细结构。当苯环上的一个氢原子或两个氢原子被其他 基团取代时,吸收带波长将发生变化。除个别取代基外, 绝大多数取代基都能使吸收带红移,E1带将移动185-220n m、E2带将移到205-250nm、B带将移到260-290nm。当 取代基含有n电子时,则在275-330nm范围将出现R吸收带。
O RC-R1
C-C、C=O、C-H
σ→σ* 、π→π*、 n→σ* 、n →π*
乙醛 290nm、丙酮280nm, 吸收强度较弱,εmax≤100,
3、醇、醚、含氮、含硫化合物及卤化物
A、醇、醚 ROH ROR1
σ→σ* 、n→σ*
紫外-可见吸收光谱与红外光谱
紫外-可见吸收光谱与红外光谱基本概念紫外-可见吸收光谱:让不同波长的光通过待测物,经待测物吸收后,测量其对不同波长光的吸收程度(吸光度A),以吸光度A为纵坐标,辐射波长为横坐标作图,得到该物质的吸收光谱或吸收曲线,即为紫外—可见吸收光谱。
红外光谱:又称为分子振动转动光谱,属分子吸收光谱。
样品受到频率连续变化的红外光照射时,分子吸收其中一些频率的辐射,分子振动或转动引起偶极矩的净变化,使振-转能级从基态跃迁到激发态,相应于这些区域的透射光强减弱,记录百分透过率T%对波数或波长的曲线,即为红外光谱。
两者都是红分了的吸收光谱图。
区别--起源不同1.紫外吸收光谱由电子能级跃迁引起紫外线波长短、频率高、光子能量大,能引起分子外层电子的能级跃迁。
电子跃迁虽然伴随着振动及转动能级跃迁,但因后者能级差小,常被紫外曲线所淹没。
除某些化合物蒸气(如苯等)的紫外吸收光谱会显现振动能级跃起迁外,一般不显现。
因此,紫外吸收光谱属电子光谱。
光谱简单。
2.中红外吸收光谱由振—转能级跃迁引起? 红外线的波长比紫外线长,光子能量比紫外线小得多,只能收起分子的振动能级并伴随转动能级的跃迁,因而中红外光谱是振动—转动光谱,光谱复杂。
适用范围紫外吸收光谱法只适用于芳香族或具有共轭结构的不饱和脂肪族化合物及某些无物的定性分析,不适用于饱和有机化合物。
红外吸收光谱法不受此限,在中红外区,能测得所有有机化合物的特征红外光谱,用于定性分析及结构研究,而且其特征性远远高于紫外吸收光谱,除此之外,红外光谱还可以用于某些无机物的研究。
紫外分光光度法测定对象的物态以溶液为主,以及少数物质的蒸气;而红外分光光度法的测定对象比紫外分光光度法广泛,可以测定气、液、固体样品,并以测定固体样品最为方便。
红外分光光度法主要用于定性鉴及测定有机化合物的分子结构,紫外分光光度法主要用于定量分析及测定某些化合物的类别等。
特性红外光谱的特征性比紫外光谱强。
因为紫外光谱主要是分子的∏电子或n电子跃迁所产生的吸收光谱。
紫外可见光谱
-胡罗卜素
咖啡因 阿斯匹林
几种有机化合
物的分子吸收光 谱图。
丙酮
2、分子吸收光谱跃迁类型 可能的跃迁类型
有机分子包括:成键轨道:、 ;反键轨道:*、*;非键轨道:n
••••C••
o
O
o
o o
•= =
o=n
各轨道能级高低顺序: n**;
可能的跃迁:-*;-*;-*;n-*;-*;n-*
(3)含有杂原子的有机化合物
杂原子(O、N 、S、Cl等)上有未成键的电子容易被激发产生n—*
n—π*跃迁。
分子可产生的跃迁:
n—π*、n—*、 π—π*、π—*、 —π*、 —*
-*,-*,-* 和n-* 跃迁能量较高,跃迁产生的吸收
谱位于真空紫外区,在此不加讨论。
-*和n-*两种跃迁的能量小,相应波长出现在近紫外区甚
助色团(Auxochromous group) :
含有孤对电子,可使生色团吸收峰移动并 提高吸收强度的一些官能团,称之为助色团。
红移或蓝移(Redshift or blueshift):
在分子中引入的一些基团或受到其它外界因素影响, 吸收峰向长波方向(红移)或短波方向(蓝移)移动的 现象。
分子发生红移或蓝移的因素:
(1)饱和烃类化合物
饱和烃类分子中只含有б健电子,因此只能产生 --- *跃迁。 —*:C—H共价键,如CH4(125nm);
C—C键,如C2H6 (135nm),处于真空紫外区。
(2)不饱和烃类化合物
不饱和烃类分子中既有 键电子,又有π键电子,其电子能级图如图所示。
—* 和—*跃迁:所需能量比—*跃迁能量小,波长处于真空紫外区。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
紫外可见吸收光谱计算是通过分析样品在紫外可见光谱范围内的吸收特性来确定其化学成分或浓度的方法。
下面是一般的紫外可见吸收光谱计算的基本步骤:
获取吸光度数据:使用紫外可见光谱仪器对待测样品进行扫描,记录下各个波长处的吸光度值。
确定基线:分析样品的吸光度数据中可能存在基线漂移或背景吸光度的影响。
通过选择不含待测物的溶剂或参考物质,测量其吸光度作为基线。
在后续计算中,需要减去基线吸光度以消除背景影响。
确定吸收峰位置:根据吸光度数据,确定待测物在紫外可见光谱中的吸收峰位置。
吸收峰通常对应于特定化合物或化学基团的特征吸收波长。
计算浓度或测定化学成分:根据已知的吸光系数和比尔定律(Beer-Lambert Law),利用吸光度数据计算待测物的浓度或测定样品中化学成分的含量。
比尔定律表示了吸光度与浓度之间的线性关系,即A = εcl,其中A为吸光度,ε为摩尔吸光系数,c为溶液中的物质浓度,l 为光程长度。
数据处理和解释:根据实际需求,对吸光度数据进行进一步处理和解释。
这可能包括绘制吸收光谱曲线、比较不同样品的吸收特征、构建校准曲线等。
需要注意的是,在进行紫外可见吸收光谱计算时,需要注意选择合适的波长范围、溶剂、光程长度和合适的校准曲线等,以确保测量的准确性和可靠性。
此外,根据待测物的特性和实验条件,还可能需要进行附加的样品预处理、峰识别和数据分析等步骤。