临床试验项目管理PPT课件
合集下载
药物临床试验管理制度和流程ppt课件

➢取得伦理批件后,申办者/CRO、机构办公室与研究 者起草合同
➢最后由申办者/CRO与机构主任签字生效。
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
步骤七:接爱临床试验材料及药物
申办者/CRO将临床试验材料交机构办公室,经办公 室备份后分发项目研究小组
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
申办者提供药物与临床试验药物相关的资料 专业科室的软硬件设施要达到要求,研究人员要
有资格并经过培训 研究者须对受试者在医疗上应认真负责 必须严格按照试验方案开展试验 研究者须保留试验过程中的源文件和源数据;将
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
国家食品药品监督管理局认定的专业才能开展 要有充分的科学依据,并《赫尔辛基宣言》和《人
体生物医学研究国际道德指南》规定的原则 申办者必须出具国家食品药品监督管理局的批件及 药检部门的检测报告 药物临床试验必须经过伦理委员会批准后方可进行 受试者必须自愿,并签署知情同意书
为参加单位,PI组织研究人员进行项目总结。 申办者/CRO将总结报告交至机构办公室,待“临
床试验项目结题签认表”签字确定后,由机构主 任会议审议、签字、盖章
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
➢最后由申办者/CRO与机构主任签字生效。
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
步骤七:接爱临床试验材料及药物
申办者/CRO将临床试验材料交机构办公室,经办公 室备份后分发项目研究小组
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
申办者提供药物与临床试验药物相关的资料 专业科室的软硬件设施要达到要求,研究人员要
有资格并经过培训 研究者须对受试者在医疗上应认真负责 必须严格按照试验方案开展试验 研究者须保留试验过程中的源文件和源数据;将
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
国家食品药品监督管理局认定的专业才能开展 要有充分的科学依据,并《赫尔辛基宣言》和《人
体生物医学研究国际道德指南》规定的原则 申办者必须出具国家食品药品监督管理局的批件及 药检部门的检测报告 药物临床试验必须经过伦理委员会批准后方可进行 受试者必须自愿,并签署知情同意书
为参加单位,PI组织研究人员进行项目总结。 申办者/CRO将总结报告交至机构办公室,待“临
床试验项目结题签认表”签字确定后,由机构主 任会议审议、签字、盖章
在整堂课的教学中,刘教师总是让学 生带着 问题来 学习, 而问题 的设置 具有一 定的梯 度,由 浅入深 ,所提 出的问 题也很 明确
临床药物试验运行管理制度
药品临床试验管理规范ppt课件

8.
9.
研究中的药品应按GMP要求生产、处理与保存, 并应按照批准了的试验方案中的规定使用。
能确保临床试验各方面质量的具有操作规程的 工作系统应加以实施。
ICH指导原则(ICH-GCP)
• • • • • • • • • 内容目录 前言 1. 专业术语名词解释 2. ICH-GCP基本原则 3. 机构审查委员会/独立的伦理委员会 3.1 职责 3.2 组成,功能与操作 3.3 工作程序 3.4 记录
ichichgcp31职责32组成功能与操作33工作程序34记录ichgcp续141研究者的资格与认同42适当的研究条件43对受试者的医疗责任44与irbiec对话45对试验方案的依从性46试验药品47随机化步骤或试验中失盲48受试者知情同意书49记录与报告410进展报告411安全性报告412提前终止试验与试验暂停413试验研究者的总结报告ichgcp51确保质量与质量控制52合同研究组织cro53医学专长54试验设计55试验管理数据处理与记录保管56试验研究者选择57现有与试验有关的责任与功能的分配58对受试者与研究者的补偿59试验费用510向管理当局的报告提交书511irbiec审批件512有关研究中药品的信息资料513研究中药品的制造包装标签与编码ichgcp514研究中药品的供应与管理515记录通路516安全性情报517药物不良反应报告518监视monitoring5181目的5182监视员的选择与资格5183监视范围与性质5184监视员职责5185监视程序5186监视报告ichgcp519监察audit5191目的5192监察人员的选择与资格5193监察程序520缺乏依从性521提前终止或试验暂停522临床试验研究报告523多中心临床试验ichgcp续561一般资料信息62背景资料信息63试验目的与本项试验目的64试验设计65受试者的选择与剔除66受试者的治疗67疗效评价68安全性评价69统计分析ichgcp610原始资料文件的直接通路611质量控制与确保质量的程序612伦理考虑613数据处理与记录保管614经费与保险615发表政策616增补ichgcp续771简介721标题页
9.
研究中的药品应按GMP要求生产、处理与保存, 并应按照批准了的试验方案中的规定使用。
能确保临床试验各方面质量的具有操作规程的 工作系统应加以实施。
ICH指导原则(ICH-GCP)
• • • • • • • • • 内容目录 前言 1. 专业术语名词解释 2. ICH-GCP基本原则 3. 机构审查委员会/独立的伦理委员会 3.1 职责 3.2 组成,功能与操作 3.3 工作程序 3.4 记录
ichichgcp31职责32组成功能与操作33工作程序34记录ichgcp续141研究者的资格与认同42适当的研究条件43对受试者的医疗责任44与irbiec对话45对试验方案的依从性46试验药品47随机化步骤或试验中失盲48受试者知情同意书49记录与报告410进展报告411安全性报告412提前终止试验与试验暂停413试验研究者的总结报告ichgcp51确保质量与质量控制52合同研究组织cro53医学专长54试验设计55试验管理数据处理与记录保管56试验研究者选择57现有与试验有关的责任与功能的分配58对受试者与研究者的补偿59试验费用510向管理当局的报告提交书511irbiec审批件512有关研究中药品的信息资料513研究中药品的制造包装标签与编码ichgcp514研究中药品的供应与管理515记录通路516安全性情报517药物不良反应报告518监视monitoring5181目的5182监视员的选择与资格5183监视范围与性质5184监视员职责5185监视程序5186监视报告ichgcp519监察audit5191目的5192监察人员的选择与资格5193监察程序520缺乏依从性521提前终止或试验暂停522临床试验研究报告523多中心临床试验ichgcp续561一般资料信息62背景资料信息63试验目的与本项试验目的64试验设计65受试者的选择与剔除66受试者的治疗67疗效评价68安全性评价69统计分析ichgcp610原始资料文件的直接通路611质量控制与确保质量的程序612伦理考虑613数据处理与记录保管614经费与保险615发表政策616增补ichgcp续771简介721标题页
药物临床试验质量管理制度PPT(16张)

•
19、大家常说一句话,认真你就输了,可是不认真的话,这辈子你就废了,自己的人生都不认真面对的话,那谁要认真对待你。
•
20、没有收拾残局的能力,就别放纵善变的情绪。
•
1、不是井里没有水,而是你挖的不够深。不是成功来得慢,而是你努力的不够多。
•
2、孤单一人的时间使自己变得优秀,给来的人一个惊喜,也给自己一个好的交代。
•
13、认识到我们的所见所闻都是假象,认识到此生都是虚幻,我们才能真正认识到佛法的真相。钱多了会压死你,你承受得了吗?带,带不走,放,放不下。时时刻刻发悲心,饶益众生为他人。
•
14、梦想总是跑在我的前面。努力追寻它们,为了那一瞬间的同步,这就是动人的生命奇迹。
•
15、懒惰不会让你一下子跌倒,但会在不知不觉中减少你的收获;勤奋也不会让你一夜成功,但会在不知不觉中积累你的成果。人生需要挑战,更需要坚持和勤奋!
GCP的重要作用
GCP可以保证:
1. 临床研究中,受试者得到适当的保护; 2. 所有的研究机构均具有良好的科学依据,设计合理; 3. 研究过程规范合理,记录完整、真实,分析结果可靠; 4.提高临床研究的质量; 5. 有利于管理部门的监督; 6. 有利于提高医疗水平,扩展临床医学知识。
GCP的重要作用
•
1、想要体面生活,又觉得打拼辛苦;想要健康身体,又无法坚持运动。人最失败的,莫过于对自己不负责任,连答应自己的事都办不到,又何必抱怨这个世界都和你作对?人生的道理很简单,你想要什么,就去付出足够的努力。
•
2、时间是最公平的,活一天就拥有24小时,差别只是珍惜。你若不相信努力和时光,时光一定第一个辜负你。有梦想就立刻行动,因为现在过的每一天,都是余生中最年轻的一天。
临床试验项目管理培训课件
1/10/2021
临床试验1项3 目管理
文件管理(3)
试验结束阶段
试验用药的回收和销毁 CRF的回收及数据的质疑 试验文件完整性的确认 伦理委员会的通报 临床试验的报告
1/10/2021
临床试验1项4 目管理
临床试验项目管理要素
文件管理
进度管理
质量管理 风险管理 1/10/2021
临床试验1项5 目管理
Have blood pressure and heart/pulse rate been taken at this visit?
No
Yes
Please record the results in the Vital Signs Log at the
end
of the CRF.
Is there any clinically significant abnormality?
病史(患病时间等) 目前身体状况、伴随疾病和用药 近期停止用药时间(满足方案要求) 受试者参加临床试验号/签署知情同意书 访视日期 实验室/X-线等结果 试验用药数量及伴随治疗记录
1/10/2021
临床试验9项目管理
1/10/2021
临床试验1项0 目管理
Protocol 265805/249
批文(CFDA批文、伦理委员会批件及其成员) 签字合同(研究者合同、财务规定) 试验器械药品器械接收单和检测报告 实验室文件(正常值范围、质控证明) 研究者简历、研究者授权表及相关文件
1/10/2021
临床试验5项目管理
临床试验方案:
方案的形成过程中需申办者 与研究者讨论,并严格遵守 中国注册法规要求。
计划阶段 撰写方案摘要 选择研究者
临床试验项目管理医学PPT课件

临床试验项目管理初探
1
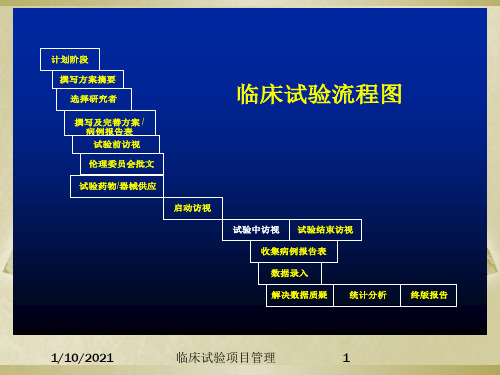
计划阶段 撰写方案摘要 选择研究者 撰写及完善方案 / 病例报告表 试验前访视 伦理委员会批文 试验药物/器械供应 启动访视 试验中访视 试验结束访视
临床试验流程图
收集病例报告表 数据录入 解决数据质疑 统计分析 终版报告
2
临床试验项目的基本要素
产品
(临床试验报告)
时间表
资源
• 方案+CRF • 参与人员 • 机构口碑
21
质量管理(2)-质量就是生命
试验进行阶段质量管理:
• 监察 • 稽查 • CRC
22
• 确认受试者符合入组标准; • 确认试验按试验方案进行所有访视;
• 随时纠正违背方案的事件。
23
质量管理(3)-质量就是生命
试验结束阶段质量管理:
• 数据和统计
VITAL SIGNS Weight (without shoes) (kg) Sitting Blood Pressure and Heart/Pulse Rate Prior to any blood pressure measurements being taken, subjects should have rested in the clinic for at least 5 minutes. Blood pressure must be measured in the same arm at each visit. Have blood pressure and heart/pulse rate been taken at this visit? No end of the CRF. Is there any clinically significant abnormality? No Yes Arm : please record details in Significant Medical/Surgical History page. Left Right Yes Please record the results in the Vital Signs Log at the Height (m)
1
计划阶段 撰写方案摘要 选择研究者 撰写及完善方案 / 病例报告表 试验前访视 伦理委员会批文 试验药物/器械供应 启动访视 试验中访视 试验结束访视
临床试验流程图
收集病例报告表 数据录入 解决数据质疑 统计分析 终版报告
2
临床试验项目的基本要素
产品
(临床试验报告)
时间表
资源
• 方案+CRF • 参与人员 • 机构口碑
21
质量管理(2)-质量就是生命
试验进行阶段质量管理:
• 监察 • 稽查 • CRC
22
• 确认受试者符合入组标准; • 确认试验按试验方案进行所有访视;
• 随时纠正违背方案的事件。
23
质量管理(3)-质量就是生命
试验结束阶段质量管理:
• 数据和统计
VITAL SIGNS Weight (without shoes) (kg) Sitting Blood Pressure and Heart/Pulse Rate Prior to any blood pressure measurements being taken, subjects should have rested in the clinic for at least 5 minutes. Blood pressure must be measured in the same arm at each visit. Have blood pressure and heart/pulse rate been taken at this visit? No end of the CRF. Is there any clinically significant abnormality? No Yes Arm : please record details in Significant Medical/Surgical History page. Left Right Yes Please record the results in the Vital Signs Log at the Height (m)
《临床试验项目管理》课件

3
制定计划
根据实际情况,制定多部门间的相关计划和协调方案,实现多部门间的协同合作 和管理。
临床试验项目管理的未来发展趋势
技术区块链技术将应 用到临床研究中
利用区块链技术建立查处临床 研究中的数据造假问题,促进 临床研究的透明与诚信。
经费
经费预算和使用方面,更加注 重效率和利用率,采用数字记 账和纪录等技术提升经费的评 估和审批效率。
时间分配
合理安排试验时间表,建立时间点监控机制,及时调整计划,保持试验进度和效果的协调性。
执行监督
细化制定实施规范、流程、操作手册,加强现场监督,并及时反馈和汇报试验计划、时间表 和执行情况。
预算和资源管理
预算管理
合理制定预算,降低费用,控 制风险,最终保证试验的顺利 进行和预算的有效利用。
人力资源
风险许可
采用风险、不确定和时间的综 合考虑方法,及时评估和批准 新样本,为临床试验研究提供 更高效和科学的统筹管理。
管理人员的职业道路和发展方向
1 进修学习
通过科目考试、技能考核等方式提升自己的专业背景和管理技能,培养和提升自己的管 理层次。
2 现场培训
通过参加项目、实验安排等现场实训,在实践中不断进取、提高实际操作能力和经验。
试验材料和设备的采购和管理
试验材料 药品 仪器设备 实验材料
采购和管理需求
统一保管、分类存储,符合GMP/GCP的标准, 保证药品的质量可控。
检查、维护及运输设备,保证仪器设备的整体性、 精准性和可用性。
标明采集时间、采集者等数据,建立实验材料样 本库,确保数据的可追溯性和与后续数据的高度 可比性
3 内部审核
机构内部根据规定要求进行内部审核,确保临床试验项目管理流程及时产生审查、申报 和内部审核文件。
临床试验项目培训---药物临床试验质量管理规范(GCP)ppt课件

在一名集中营囚犯身上诱导低温
减压实验,杀死了一名囚犯
9
Hale Waihona Puke 相关国际法规的发展历程-6
1947 年的纽伦堡审判
骇人听闻的纳粹实验暴露给公众 23 名纳粹医生 受到了战争罪行的审判
一名波兰证人在法庭展示了气性坏 疽实验给腿部留下的伤疤
纳粹医生 Karl Brandt 被 判处死刑
10
相关国际法规的发展历程-7
的概念
1978 年 , Belmont 报告(美国国家委员会为保护参加生物医学和 行为学研究的人体实验对象而制定的道德原则和准则)
1974 年,美国国会任命了一个国家委员会,以保护参加临 床研究的受试者
审核临床研究的伦理原则 :
自主性 受益性 公正性
16
相关国际法规的发展历程-13
念; FDA 提出了临床试验质量管理规范的概念; ICH 提供了临床研究中的全球性指导原则。
21
我国新药管理与GCP发展概况
22
我国新药管理与GCP发展概况-1
1963年卫生部、化工部、商业部《关于药政管理的若干规定》 是我国最早关于药品临床试验的规定; 1965年卫生部、化工部《药品新产品管理暂行规定》; 1978年国务院《药政管理条例》; 1979年卫生部《新药管理办法》; 1985年《中华人民共和国药品管理法》; 1985年卫生部《新药审批办法》标志我国的新药管理进入法制 化时期; 1988年卫生部《关于新药审批管理若干补充规定》,同时颁发 了15类药物的临床试验指导原则; 1992年卫生部《关于药品审批管理若干问题的通知》。
公众到美国国会群起抗议示威; 1906 年,通过 “完全食品与药物法 ”,食品与
药品管理局 (FDA) 诞生; 要求每种药物的标签必须准确; 未要求检测安全性。
药物临床试验管理规范培训ppt课件

❖ 要求依据充分,操作性强,要求清晰准确, 要求格式统一,具有可修订性。
精品课件
药物临床试验最低病例数(试验组) 为多少 ?
❖ Ⅰ期临床试验-人体药理学(药代动力学和最大耐 受性研究)20~30例;
❖ Ⅱ期临床试验-100例; ❖ Ⅲ期临床试验-300例; ❖ Ⅳ期临床试验-临床应用2000例; ❖ 生物利用度试验19~25例。 注:题中所示病例数为试验组病例数,对照组病例数
精品课件
新药分类 ?
❖ 共分6类,1-2类是国内外均未上市的药品,3 类是国外已上市但国内未上市的药品,4-5类 是改酸根、碱基,改剂型的药品,6类是纯仿 制药。
精品课件
❖ 1、未在国内外上市销售的药品: ❖ 2、改变给药途径且尚未在国内外上市销售的制剂。 ❖ 3、已在国外上市销售但尚未在国内上市销售的药品
❖ (3)新药临床前药理学、毒理学、动物体内药效动学及药 动学的研究。
❖ (4)在人体内的药动学、安全性及疗效以及市场应用经验 的研究,如注明己上市的国家以及所有上市后累计经验的重 要资料(如处方、剂量、用法和不良反立等)。
研究者手册是新药资料概要,可作为研究者指南,
向研究者提供对新药可能出现的危险、药物过量和不良反应
精品课件
我国对临床试验的资料保存时间有何 规定 ?
❖ 研究者— 临床试验结束后5年 ❖ 申办者— 试验药物获准上市后5年
精品课件
监查、稽查和视察有何不同 ?
❖ 监查(monitoring):指临床试验申办单位委派人 员对临床试验情况进行检查,以保证临床试验数据 的质量并保护受试者的安全和合法权益。
❖ ②等效性(equivalence)检验:确认两种或多种治 疗的效果差别大小在临床上并无重要意义,即试验 药与阳性对照在疗效上相当。
精品课件
药物临床试验最低病例数(试验组) 为多少 ?
❖ Ⅰ期临床试验-人体药理学(药代动力学和最大耐 受性研究)20~30例;
❖ Ⅱ期临床试验-100例; ❖ Ⅲ期临床试验-300例; ❖ Ⅳ期临床试验-临床应用2000例; ❖ 生物利用度试验19~25例。 注:题中所示病例数为试验组病例数,对照组病例数
精品课件
新药分类 ?
❖ 共分6类,1-2类是国内外均未上市的药品,3 类是国外已上市但国内未上市的药品,4-5类 是改酸根、碱基,改剂型的药品,6类是纯仿 制药。
精品课件
❖ 1、未在国内外上市销售的药品: ❖ 2、改变给药途径且尚未在国内外上市销售的制剂。 ❖ 3、已在国外上市销售但尚未在国内上市销售的药品
❖ (3)新药临床前药理学、毒理学、动物体内药效动学及药 动学的研究。
❖ (4)在人体内的药动学、安全性及疗效以及市场应用经验 的研究,如注明己上市的国家以及所有上市后累计经验的重 要资料(如处方、剂量、用法和不良反立等)。
研究者手册是新药资料概要,可作为研究者指南,
向研究者提供对新药可能出现的危险、药物过量和不良反应
精品课件
我国对临床试验的资料保存时间有何 规定 ?
❖ 研究者— 临床试验结束后5年 ❖ 申办者— 试验药物获准上市后5年
精品课件
监查、稽查和视察有何不同 ?
❖ 监查(monitoring):指临床试验申办单位委派人 员对临床试验情况进行检查,以保证临床试验数据 的质量并保护受试者的安全和合法权益。
❖ ②等效性(equivalence)检验:确认两种或多种治 疗的效果差别大小在临床上并无重要意义,即试验 药与阳性对照在疗效上相当。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
临床试验项目管理初探
南京西格玛医药技术有限公司
刘金波
计划阶段 撰写方案摘要 选择研究者
撰写及完善方案 / 病例报告表 试验前访视
伦理委员会批文
试验药物/器械供应 试验结束访视
收集病例报告表
数据录入
解决数据质疑
统计分析
终版报告
临床试验项目的基本要素
时间表
产品
(临床试验报告)
病例至少应包括的内容:
病史(患病时间等) 目前身体状况、伴随疾病和用药 近期停止用药时间(满足方案要求) 受试者参加临床试验号/签署知情同意书 访视日期 实验室/X-线等结果 试验用药数量及伴随治疗记录
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
SUCCESS
THANK YOU
2020/12/9
文件管理(3)
试验结束阶段
试验用药的回收和销毁 CRF的回收及数据的质疑 试验文件完整性的确认 伦理委员会的通报 临床试验的报告
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
原始文件是指原始的资料、数据和记录。 病历 实验室报告 受试者日记 药品发放记录等
原始文件用于证明临床试验数据的真实性、准确、可靠的证据。 满足方案要求 提供所有CRF要求的数据 原则上,是不容改动的
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
知情同意书是临床试验的重要文 件,怎样强调也不过分!
知情同意书过程也重要。
受试者参加临床试验前应签署知 情同意书,所以受试者签署姓名 和时间是非常重要的文字证明。
知情同意书时间至少与访视1同 日。
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
Prior to any blood pressure measurements being taken, subjects should have rested in the clinic for at least 5 minutes. Blood pressure must be measured in the same arm at each visit.
更改有原始数据支持 在错误上划横杠、旁加正确内容 研究者签署姓名和日期
082 086 SXC
25/2/04
监查员确保CRF与原始资料相一致
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
文件管理(2)
临床试验进行阶段: 更新文件 填写后的知情同意书、CRF 严重不良事件报告及原始文件 受试者筛选表登记表 试验药品管理记录表
资源
临床试验项目的生命周期
立项
计划(试验前)
实施(试验中)
结束(试验后)
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
临床试验项目管理要素
文件管理
进度管理 质量管理 风险管理
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
No
Yes
please record details in Significant Medical/Surgical History page.
Arm :
Left
Right
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
Protocol 265805/249
Centre Number
Subject Number
Subject Initials
Screening
Visit Date DD MM YY
Have blood pressure and heart/pulse rate been taken at this visit?
No
Yes
Please record the results in the Vital Signs Log at the
end
of the CRF.
Is there any clinically significant abnormality?
临床试验方案:
方案的形成过程中需申办者 与研究者讨论,并严格遵守 中国注册法规要求。
方案必须经过伦理委员会书 面批准。
方案终稿需有研究者签名, 并声明遵循方案实施该试验。
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
DEMOGRAPHY
Date of Birth
DD MM YY
Race:
White
Black
Sex
Male
Female
Chinese
Other: specify:
VITAL SIGNS
Weight (without shoes)
(kg)
Height (m)
•
•
Sitting Blood Pressure and Heart/Pulse Rate
文件管理(1)
试验前文件内容:
试验文件(研究者手册、签字方案、病例报告表、 知情同意书)
批文(CFDA批文、伦理委员会批件及其成员) 签字合同(研究者合同、财务规定) 试验器械药品器械接收单和检测报告 实验室文件(正常值范围、质控证明) 研究者简历、研究者授权表及相关文件
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
南京西格玛医药技术有限公司
刘金波
计划阶段 撰写方案摘要 选择研究者
撰写及完善方案 / 病例报告表 试验前访视
伦理委员会批文
试验药物/器械供应 试验结束访视
收集病例报告表
数据录入
解决数据质疑
统计分析
终版报告
临床试验项目的基本要素
时间表
产品
(临床试验报告)
病例至少应包括的内容:
病史(患病时间等) 目前身体状况、伴随疾病和用药 近期停止用药时间(满足方案要求) 受试者参加临床试验号/签署知情同意书 访视日期 实验室/X-线等结果 试验用药数量及伴随治疗记录
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
SUCCESS
THANK YOU
2020/12/9
文件管理(3)
试验结束阶段
试验用药的回收和销毁 CRF的回收及数据的质疑 试验文件完整性的确认 伦理委员会的通报 临床试验的报告
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
原始文件是指原始的资料、数据和记录。 病历 实验室报告 受试者日记 药品发放记录等
原始文件用于证明临床试验数据的真实性、准确、可靠的证据。 满足方案要求 提供所有CRF要求的数据 原则上,是不容改动的
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
知情同意书是临床试验的重要文 件,怎样强调也不过分!
知情同意书过程也重要。
受试者参加临床试验前应签署知 情同意书,所以受试者签署姓名 和时间是非常重要的文字证明。
知情同意书时间至少与访视1同 日。
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
Prior to any blood pressure measurements being taken, subjects should have rested in the clinic for at least 5 minutes. Blood pressure must be measured in the same arm at each visit.
更改有原始数据支持 在错误上划横杠、旁加正确内容 研究者签署姓名和日期
082 086 SXC
25/2/04
监查员确保CRF与原始资料相一致
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
文件管理(2)
临床试验进行阶段: 更新文件 填写后的知情同意书、CRF 严重不良事件报告及原始文件 受试者筛选表登记表 试验药品管理记录表
资源
临床试验项目的生命周期
立项
计划(试验前)
实施(试验中)
结束(试验后)
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
临床试验项目管理要素
文件管理
进度管理 质量管理 风险管理
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
No
Yes
please record details in Significant Medical/Surgical History page.
Arm :
Left
Right
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
Protocol 265805/249
Centre Number
Subject Number
Subject Initials
Screening
Visit Date DD MM YY
Have blood pressure and heart/pulse rate been taken at this visit?
No
Yes
Please record the results in the Vital Signs Log at the
end
of the CRF.
Is there any clinically significant abnormality?
临床试验方案:
方案的形成过程中需申办者 与研究者讨论,并严格遵守 中国注册法规要求。
方案必须经过伦理委员会书 面批准。
方案终稿需有研究者签名, 并声明遵循方案实施该试验。
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司
DEMOGRAPHY
Date of Birth
DD MM YY
Race:
White
Black
Sex
Male
Female
Chinese
Other: specify:
VITAL SIGNS
Weight (without shoes)
(kg)
Height (m)
•
•
Sitting Blood Pressure and Heart/Pulse Rate
文件管理(1)
试验前文件内容:
试验文件(研究者手册、签字方案、病例报告表、 知情同意书)
批文(CFDA批文、伦理委员会批件及其成员) 签字合同(研究者合同、财务规定) 试验器械药品器械接收单和检测报告 实验室文件(正常值范围、质控证明) 研究者简历、研究者授权表及相关文件
南京西格玛医药技术有限公司-一家深耕于医疗器械市场咨询、注册代理、临床试验和统计分析的专业公司