经典化学合成反应标准操作醛酮的合成
12 醛和酮化学性质

中间体 烷氧基负离子
2017/1/10 21
羟醛缩合反应机理(四步) 亲核加成-消除反应
A: 生成亲核试剂(烯醇负离子) B: 亲核加成 C: 生成β-羟醛
H3 C OC H
H2O
OH H3 C C H
CH2 CHO
CH2 CHO
D: 脱水形成不饱和醛
OH H3 C C H
,— H2O
H3C
CH
2017/1/10 30
A: 托伦斯试剂
R(Ar) CHO
+
2 Ag(NH3)2OH
RCOONH4
Байду номын сангаас
+
2 Ag
+
3 NH3
+
H2O
银镜反应 B: 费林试剂
RCHO
+
2 Cu
2+
+
OH
-
+
H2O
RCOO -
+
Cu2O
+
4H
+
2017/1/10
31
托伦斯、菲林试剂的特点
① 此两种试剂都是弱氧化剂,只氧化醛基,对原结 构中的双键没有影响;
2017/1/10 28
卤仿反应的应用:
定性鉴别
合成其它方法难以得到的、减少了一个碳的羧酸。
如:制备2,2-二甲基丙酸
嚬 呐 酮
CH3 O H3 C C CH3 C CH3
① Br2 / NaOH ② H+
H3 C
CH3 O C CH3 C OH
思考:能否用卤烃氰解的方法?
CH3 H3 C
CH2CH2
E
有机化学(主编邓苏鲁-化工第四版)课件:第9章+醛和酮-下
结束
1
甲醛易溶液于水,一般以水溶液的方式保存和出售,含37%~40%甲 醛、8%甲醇水溶液叫“福尔马林”,常用作杀菌剂和生物标本的防腐剂。 也可作农药用于防止稻瘟病。甲醛有毒,对眼黏膜、皮肤都有刺激作用,
过量吸入其蒸气会引起中毒。现代室内装饰材料用的木工板和家具等都
会不同程度的释放出有毒的甲醛,严重污染室内空气,刚使用时应注意 通风,以防中毒。 甲醛性质活泼,极易聚合。其水溶液久置或蒸发浓缩可生成直链的聚 合体——多聚甲醛— CH2O— ( ) n 。多聚甲醛为白色固体、加热至180~200 ℃时,可
应生成无色结晶,也能与碘的NaOH溶液反应。B不能与斐林试剂反应,A与浓硫酸脱 水后生成烯烃C(C3H6)。试推测A,B的构造式。
⒑ 化合物A、B、C,分子式均为C4H8O ;A、B可以和苯肼反应生成沉淀,而C不
能;B可以与斐林试剂反应,而A、C不能;A、C能发生碘仿反应,而B不能;试推 测A、B、C的构造式。
α-羟基磺酸钠
O R—C—H(R„ )
2R“OH ,干HCl
OR“ R—C—H(R„ ) OR”
NO2 NO2
2,4—二硝基苯腙 缩醛
NO2
H2N—NH— NO2
R—C—H(R„ )
12
结束
O CH3—C—H(R) )
O
3NaOI CI —C—H(R (I2—NaOH) 3
O
(R)H—C—ONa + CHI3↓
习
CH3 CHO ︱ ︱ ⑴ CH3—CH—CH2—CH—CH3 CH2—CH3 ︱ CH3—CH—CH2—CHO O ︱ ︱ ⑸ ⑻
C—CH3
题
CH3 O Cl ︱ ︱ ︱ ︱ CH3—CH—C—CH—CH3 CH3 ︱ ⑷
有机化学人名反应

有机人名反应及机理索引:Arbuzov反应Arndt-Eister反应Baeyer-Villiger 氧化Beckmann 重排Birch 还原Bischler-Napieralski 合成法Bouveault-Blanc还原Bucherer 反应Cannizzaro 反应Chichibabin 反应Claisen 酯缩合反应Claisen-Schmidt 反应Clemmensen 还原Combes 合成法Cope 重排Cope 消除反应Curtius 反应Dakin 反应Darzens 反应Demjanov 重排Dieckmann 缩合反应Elbs 反应Eschweiler-Clarke 反应Favorskii 反应Favorskii 重排Friedel-Crafts烷基化反应Friedel-Crafts酰基化反应Fries 重排Gabriel 合成法Gattermann 反应Gattermann-Koch 反应Gomberg-Bachmann 反应Hantzsch 合成法Haworth 反应Hell-V olhard-Zelinski 反应Hinsberg 反应Hofmann 烷基化Hofmann 消除反应Hofmann 重排(降解)Houben-Hoesch 反应Hunsdiecker 反应Kiliani 氰化增碳法Knoevenagel 反应Knorr 反应Koble 反应Koble-Schmitt 反应Leuckart 反应Lossen反应Mannich 反应Meerwein-Ponndorf 反应Meerwein-Ponndorf 反应Michael 加成反应Norrish I和II 型裂解反应Oppenauer 氧化Paal-Knorr 反应Pictet-Spengler 合成法Pschorr 反应Reformatsky 反应Reimer-Tiemann 反应Reppe 合成法Robinson 缩环反应Rosenmund 还原Ruff 递降反应Sandmeyer 反应Schiemann 反应Schmidt反应Skraup 合成法Sommelet-Hauser 反应Stephen 还原Stevens 重排Strecker 氨基酸合成法Tiffeneau-Demjanov 重排Ullmann反应Vilsmeier 反应Wagner-Meerwein 重排Wacker 反应Williamson 合成法Wittig 反应Wittig-Horner 反应Wohl 递降反应Wolff-Kishner-黄鸣龙反应Yurév 反应Zeisel 甲氧基测定法亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
3.3醛酮高二化学精品讲义(人教版2019选择性必修3)(原卷版)

3.3 醛酮核心素养发展目标1.通过醛基中原子成键情况的分析,了解醛类的结构特点,理解乙醛的化学性质与醛基的关系,学会醛基的检验方法。
2.了解甲醛对环境和健康的影响,关注有机化合物安全使用的问题。
考点1 醛一、乙醛的性质1.醛的概念及结构特点醛是由烃基(或氢原子)与醛基相连而构成的化合物。
醛类官能团的结构简式是—CHO,饱和一元醛的通式为C n H2n O(n≥1)或C n H2n+1CHO。
2.乙醛的结构与物理性质3.乙醛的化学性质(1)加成反应①催化加氢(还原反应)乙醛中的碳氧双键和烯烃中的碳碳双键性质类似,也能与氢气发生加成反应,化学方程式为CH3CHO+H2――→催化剂△CH3CH2OH。
②与HCN加成在醛基的碳氧双键中,由于氧原子的电负性较大,碳氧双键中的电子偏向氧原子,使氧原子带部分负电荷,碳原子带部分正电荷,从而使醛基具有较强的极性。
乙醛能和一些极性试剂例如氰化氢(HCN)发生加成反应:+H—CN―→(2-羟基丙腈)。
(2)氧化反应①可燃性乙醛燃烧的化学方程式:2CH3CHO+5O2――→点燃4CO2+4H2O。
考点梳理②催化氧化乙醛在一定温度和催化剂作用下,能被氧气氧化为乙酸的化学方程式:+O 2――→催化剂△。
③与银氨溶液反应向A 中滴加氨水,现象为先产生白色沉淀后变澄清,加入乙醛,水浴加热④与新制氢氧化铜反应A 中溶液出现蓝色絮状沉淀,滴入乙醛,加热至沸腾后,C 中溶液有砖⑤乙醛能被酸性高锰酸钾溶液、溴水等强氧化剂氧化。
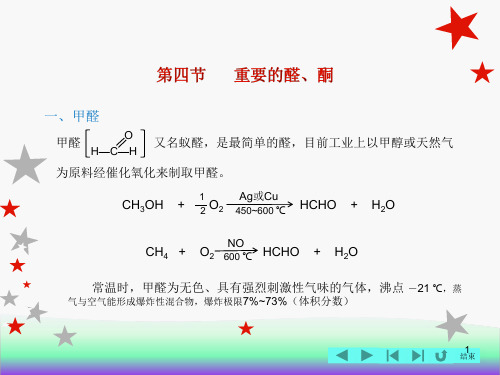
二、醛的结构与常见的醛 1.常见的醛(1)甲醛:又名蚁醛,是结构最简单的醛,结构简式为HCHO 。
通常状况下是一种无色有强烈刺激性气味的气体,易溶于水。
它的水溶液又称福尔马林,具有杀菌、防腐性能,可用于消毒和制作生物标本。
①结构特点甲醛的分子式为CH 2O ,其分子可以看成含两个醛基,如图:②甲醛氧化反应的特殊性 甲醛发生氧化反应时,可理解为――→氧化――→氧化。
醛酮缩合反应

学习情境9-4
12.2 缩合方法
主要介绍醇醛缩合或 醇酮缩合、酯缩合 12.3 应用实例。
1
缩合技术
12.1 概述
• 1、含义: • 缩合反应一般系指两个或两个以上分子间通过生 成新的碳-碳、碳-杂原子或杂原子-杂原子键,从 而形成较大的单一分子的反应。
缩合反应一般往往伴随着脱去某一种小分子化合物,如:
• 羟醛缩合反应可分为同分子醛、酮自身缩合和异分子
醛(或酮)交叉缩合两大类,在工业上都有重要用途。
3
• ① 同分子醛、酮自身缩合
• 特点:
• 是可使产物的碳链长度增加一倍,工业上可利用这种 缩合反应来制备高级醇。
O H CH3C O H OHOH CH3CH CH2C O H
H2O
CH3C
CH3CH= CHC
O O O
α 紫罗兰酮
β 紫罗兰酮
γ 紫罗兰酮
13
• 由半合成法制备紫罗兰酮:
O
缩合
O CHO CH3 C CH3 O
环化
CH CH C CH3 O
α 紫罗兰酮
β 紫罗兰酮
14
//
R R
/
C
//
O
2 R CH2OH
H
R R
/
OCH2 R C
//
H2O
OCH2 R
如:香料 1,1-二甲氧基乙烷,俗称乙醛二甲缩醛
CH3CHO 2CH3OH
H
CH3
O CH O
CH3 H2O CH3
10
(4)酯的缩合反应
• 酯缩合反应是指以羧酸酯为亲电试剂,在碱性催化剂 作用下,与含活泼氢羰基化合物的负碳离子缩合而生成β -羰基类化合物的反应,总称为克莱森(Claisen)缩合 反应。
经典合成反应标准操作

经典化学合成反应标准操作药明康德新药开发有限公司化学合成部编写、八刖有机合成研究人员在做化学反应经常碰到常规的反应手边没有现成的标准操作步骤而要去查文献,在试同一类反应时,为了寻找各种反应条件方法也得去查资料。
为了提高大家的工作效率,因此化学合成部需要一份〈经典合成反应标准操作》。
在这份材料中,我们精选药物化学中各类经典的合成反应,每类反应有什么方法,并通过实际经验对每类反应的各种条件进行点评,供大家在摸索合成条件时进行比较。
同时每种反应的标准操作,均可作为模板套用于书写客户的final report,这样可以大大节省研究人员书写final report的时间,也相应减少在报告中的文法错误。
另外本版是初版,在今后的工作中我们将根据需要修订这份材料。
药明康德新药开发有限公司化学合成部2005-6-281•胺的合成a)还原胺化b)直接烷基化c)腈的还原d)酰胺的还原e)硝基的还原f)叠氮的还原g) Hoffman 降解h)羧酸通过Cris重排2. 羧酸衍生物的合成a)酰胺化的反应b)酯化反应c)腈转化为酯和酰胺d)钯催化的插羰反应e)酯交换为酰氨3. 羧酸的合成a)醇氧化b)酯水解c)酰胺的水解d)腈的水解e)有机金属试剂的羰基化反应f)芳香甲基的氧化4. 醛酮的合成a) Weinreb酰胺合成醛酮b)醇氧化c)酯的直接还原d)有机金属试剂对腈加成合成酮5•脂肪卤代物的合成a)醇转化为脂肪溴代物通过PBr3转化通过PPh3 与CBr4转化HBr直接交换通过相应的氯代物或磺酸酯与LiBr交换、b)醇转化为脂肪氯代物通过S0CI2转化通过PPh3与CCl4转化HCl直接交换c)醇转化为脂肪碘代物通过PPh3与12转化通过相应的氯代物或磺酸酯与Nal交换6. 芳香卤代物的合成a)San dermyyer 重氮化卤代b)直接卤代c)杂环的酚羟基或醚的卤代7. 醇的合成a)羧酸或酯的还原b)醛酮的还原c)卤代烃的水解d)吡啶的氧化转位8. 酚的合成a)San dermayer 重氮化反应b)醚的水解c)Bayer-vigerlar 氧化d)硼酸的氧化9. 腈的合成a)磺酸酯或卤代烃的取代b)酰胺的脱水c)芳卤代烃的氰基取代10. 硝化反应11. 醚的合成a)芳香醚的合成酚与烷基卤代烃的直接烷基化Mitsu nobu 芳香醚化Buckwald 芳香醚化b)脂肪醚的合成醇的醚化12•脲的合成a)胺与异腈酸酯的反应b)用三光气合成脲c)羰基二咪唑(CDI)合成脲d)对硝基苯酚碳酰胺合成脲13. 烯烃的合成a) Wittig 反应b)羟基的消除c)Wittig-Horner 反应合成,-不饱和酯14. 磺酸及磺酰氯的合成a)氯磺化反应合成磺酰氯b)从硫醇合成磺酰氯c)磺化反应15. 氨基酸的合成a) Streck反应合成b)手性氨基酸的合成16. 偶联反应a) Suzuki Coupli ngb) Buckwald 芳胺化,芳酰胺化、c)Heck反应17. Mitsunobu 反应a)醇的反转b)胺的取代18. 脱羟基反应19. 酮还原为亚甲基20. 氨的保护及脱保护策略a)用碳酰胺作保护基b)苄基保护21. 醇的保护及脱保护策略a)用硅醚进行保护b)其他醚类保护22. 羧基的保护Boc 脱保护1格氏反应还原胺化卤化反应S u z u k i coupling ------------------------------------------------------------------------------------------------ - 2磺化反应n-BuLi -------------------------------------------------------------------------------------------------L i A l H 4 还原 -------------------------------------------------------------------------------------------------- 4P0CI3 的杂环氯代3水解反应-------------------------------------------------------------------------------------------- 5NaH ----------------------------------------------------------------------------------------------------___________NBS ---------------------------------------------------------------------------------------------------———————————氢化反应m-CPBA ----------------------------------------------------------------------------------------------6 EDC ---------------------------------------------------------------------------------------------------6用二光气成脲——7芳卤用n -B u L i i处理后与W e in r e b 酰胺成酮 -------------------------------------------------------------------------------------------------------------------------------------------- 7Boc上保护To a soluti on of A (2.72 g, 13.9 mmol) and tetramethylam monium hydroxide pen tahydrate (5.62 g, 31.0 mmol) in aceto nitrile (270 mL) was added di-tert-butyldicarb on ate (3.79 g; 17.4 mmol) and the resulting solution was allowed to stir 18 h at rt and concentrated. The residue was partitio ned betwee n Et2O/H2O; the phases were separated and the aqueous phase extracted twice more with Et2O. The aqueous phase was brought to pH 4 with solid citric acid and extracted with CHCI3 (3x100 mL). The orga nic extracts were comb in ed, dried (Na2SO4) andconcen trated to afford 2.58 g (63 perce nt) B as a white foam.Boc 脱保护Tert-Butyl 2-(2-methoxyphe no xy)ethylcarbamate (23.8 g, 89 mmol) in dichlorometha ne (10 ml) was cooled to 0 deg C and stirred as a mixture of trifluoroacetic acid: dichloromethane (1:1,40 ml) was added dropwise. The mixture was allowed to warm to rt, stirred for 2 hours and concen trated in vacuo. The residue was take n back up in dichlorometha ne (100 ml) and thesolutio n was washed with saturated aqueous sodium hydroge n carb on ate (3*20 ml) and aqueous sodium hydroxide (10percent, 3*20 ml), dried (Na2SO4), filtered and concentrated in vacuo to provide 2-(2-methoxyphe no xy)ethylam ine (13 g, 88perce nt yield) as a light yellow solid.Return格氏反应A stirred mixture of magn esium tur nings (23.6 g, 0.98 mol) and Et2O (200 mL) un der n itroge n is treated with a crystal of iodi ne and about 5perce nt of a soluti on of bromoetha ne (56.3 ml, 0.75 mol) in Et2O (375 mL). When the react ion starts, the rema in der of the bromoetha ne solutio n is added, dropwise at a rate sufficient to maintain a gentle reflux. After the addition, stirring is continued for 1 hour. T o this solution ofethylmagnesium bromide was slowly added a solution of 4-cya nopyridi ne (39 g, 0.375 mol) in Et2O (750 ml). The react ion mixture was warmed at reflux for 12 hours, treated with con ce ntrated H2SO4 (125 ml)/H2O (125 ml), and then washed three times with Et2O (250 ml). The aqueous portio n was made basic (PH 9) with 15perce nt NaOHReturnOsoluti on and extracted five times with 250 ml portions of Et20. The comb ined Et20 extracts were dried (MgSO4), and the solve nt was removed un der reduced pressure to afford a brow n oil (48.4 g, 95perce nt).卤化反应To a stirred solution of 8-methyl-1-nitro-naphthalene (10.6g, 56.32 mmol) and iron (III) chloride (0.45 g, 2.77 mmo) in CCI4 (150 ml) heated to 60 C was added dropwise (3.0 ml, 58.23 mmol) of bromine. After one hour, the react ion mixture was poured into saturated NaHCO3 solutio n, and the layers were separated. The aqueous layer was re-extracted with CH2CI2. The comb ined orga nic layers were dried (MgSO4) and the solve nt was removed un der reduced pressure. The crude residue was recrystallized from etha nol and the mother liquors were concen trated and then flash chromatographed on silica, eludi ng hexa nes:ethyl acetate (12: 1).ReturnReturn还原胺化 HO H 2NA solution of 2-ami no-4-ethylphe nol (1.00 g. 7.28 mmol), 2-naphthaldehyde (1.13 g, 7.28 mmol), and p-tolue nesulfo nic acid (0.05 g) in metha no I (50 ML) was stirred at room temp for 24 h. To the resultant solution, sodium borohydride (0.82 g, 22 mmol) was added in small portions. After additi on was completed, the mixture was stirred at room temperature for 30 min and concen trated un der vacuum. The residue was the n subjected to colu mn chromatography on silica gel eluted with 10percent ethyl acetate in hexane and followed by recrystallization (aqueous metha nol) yielded 450 mg (22perce nt) of an alytically pure product.Retur n+V HSuzuki coupli ngTo a mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-indole (2 g, 8.2 mnmol)and 3-bromobe nzene (0.87 ml, 8.3 mmol) in THF (28 ml) were added palladium catalystPd(PPh3)4 (284 mg, 0.25 mmol) and the freshly prepared sodium hydroxide solution (984 mg in9 ml of water).The system was degassed and the n charged with n itroge n for three times. Themixture was stirred un der n itroge n at 70 ° C oTlbiatie^i6rheoiliuti on was cooled to room temperature, diluted with ethyl acetate and separated from water layer. The ethylacetate soluti on was washed by brine, dried over Na2SO4 and concen trated. The residue waspurified on a silica gel colu mn eludi ng with hexa nes: EtOAc 9:1 to give 1.38 g (78%yield) of4-phenyl-1H-indole as a colorless liquid.Return磺化反应Chlorosulfo nic acid (4.66g, 40 mmol) is added dropwise to a cold (0 2,3-dihydro-2-trifluoroacetyl-1H-Benz[de]isoquinoline (2.9g, 8 mmol) in chloroform (800 ml).C for 30 minutes. ° The cold bath is then removed and the solution is stirred at room temperature for 1 hour thencautiously poured into ice water. The orga nic layer is separated, dried over magn esium sulfate and concen trated to afford the titlecompo und. The crude product is purified by colu mn chromatography eluted with 10% acetic ether in petroleum ether (2.36 g, 81% yield).酯化反应A mixture of 4-hydroxymethy In aphthoic acid (10 g, 50 mmol), metha no I (300 ml), and concen trate H2SO4 (2 ml) was refluxed overni ght. The in solubles were filtered off and the filtrate was concen trated. The residue was take n up in ethyl acetate and washed with aqueous NaHCO3 (2*), brine, dried over MgSO4, and concen trated to give a yellow oil. Silica gel colu mn chromatography using ethyl acetate/hexa ne (1/3) gave the desired product as a yellow oil (3.3 g,35%yield).Retur nC) solutio n ofThe result ing soluti on is stirred at 0HOHO水解反应sodium hydroxide (35ml) in tetrahydrofura n (130ml) was stirred un der reflux for 18 hours. The mixture was n eutralised using 2N hydrochloric acid, and extracted with dichlorometha ne (3x).The comb ined orga nic soluti ons were dried (MgSO4), and evaporated un der reduced pressure. The crude product was purified by column chromatography on silica gelusing an gradient of dichloromethane: methanol (100:0 to 97:3) to afford the title compound as a solid (3.11g,47.8%yield).硝化反应NO2To a cold (0 °C) suspension of 1-methylnaphthalene (5 g, 35.2 mmol) in HNO3 was added H2SO4 (5 ml) dropwise. After stirri ng the react ion for one hour, the soluti on was diluted with ethyl acetate and washed with water (3*), aqueous saturated NaHCO3 (2*) and brine, dried over MgSO4, and concen trated. The product was purified by silica gel colu mn chromatography using ReturnA solution of 1-Methyl-naphthalene-2-carboxylicacid methyl ester (7.20g, 35mmol) and 2NelutionRetur nOHethyl acetate: hexa ne (5: 95) and recrystallized from metha nol to give yellow n eedles (0.22g, 33% yield).n-BuLiTo a dry three-n ecked roun d-bottomed flask with an additi on funnel and at -78 inertatmosphere was charged with an hydrous THF (500 ml). A soluti on of n-butyllithium (2.5 Min hexane, 88 ml, 220 mmol) was added dropwise followed by addition of a solution of aceton itrile (10.43 ml, 200 mmol) in an hydrous THF (100 ml). The in ternal temperature wasmaintained below -70 °C duri ng the en tire additi on process. After 2 hr at -78 °C a soluti on of Trifluoro-acetic acid ethyl ester (14.2 g, 100 mmol) in an hydrous THF (30 ml) was added dropwise and the mixture was stirred for 1.5 hr. T o the mixture was added acetic an hydride to que nch the react ion. The reacti on mixture was allowed to warm up to rt. A precipitate was filtered and the filtrate was concentrated to give a brown oil, which was used in the next step withoutpurificatio n.ReturnLiAlH4还原A solution of 2,3-naphthale nedicarboxylic acid (4.6 g, 0.023 mole) in dry THF (135 ml, warmed toReturn°C under O50 ° to maintain solution) is added dropwise over 15 minutes to a 1.15 M lithium aluminum hydride solution in THF (45 ml, 0.052 mole). The solution is stirred 3 hours after which TLC indicated consumption of diacid and formation of a new major product. The reaction is quenched carefully with THF-water, then 2N hydrochloric acid (40 ml) is added, and the resulting mixture is extracted 3 times with ether. The comb ined ether extracts are washed with water (2 times), with saturated sodium bicarb on ate soluti on (1 time), with water, and are dried (sodium sulfate), filtered, and concentrated to give a tan solid (3.67 g). The solid is recrystallized from ethyl acetate giving the title compound (2.91 g, 67.3%yield) as a light tan crystalline material.Retur n POCI3的杂环氯代HO ClTo a suspension of 2,4-dihydroxy-5,6-dimethylpyrimidine (6.2 g, 0.044 mol) in POCl3 (25 ml) wasslowly added N,N-dimethyla nili ne (6.18 ml, 0.049 mol). The mixture was then refluxed at 125 Cfor 3 hours. After this time, the starti ng material completely dissolved in dicat ing that the reactio n was completed. The react ion mixture was cooled and the n poured slowly onto ice to que nchthe POCl3 (cauti on[ exothermic]). A precipitate formed, which was filtered and washed withice-cold water. The precipitate was dried un der high vacuum overni ght to yield2,4-dichloro-5,6-dimethyl-pyrimidine (7.2 g, 0.041 mol, 92%yield) as a yellow solid.Retur nNaHSodium hydride (50% in min eral oil, 5.5 g, 0.11 mol) was added porti on wise at 0 nitrogen atmosphere to a solution of 2-aminobenzenethiol (12 ml, 0.1 mol) in DMF (120 ml).After 0.5 h, ben zyl chloride (11.5 ml, 0.1 mol) in DMF (80 ml) was added in 0.5 h. The solutionwas stirred for 3 h while the temperature was allowed to rise to rt, then it was poured into ice/water (1000 g). The precipitate was filtered, dissolved in ethyl acetate and washed with brine. Theorga nic layer was dried over Na2SO4 and evaporated. The solid obta ined was ground in pentane (19.3 g, 90% yield).NBSA mixture of 2,4-Dichloro-6-ethyl-5-fluoro-pyrimid ine (27.46 gand n-bromosucci nimide (27.02 g , 0.152mol) in CH2Cl2 (170 ml) wasrefluxed un der a nitroge natmosphere for 36 h. Then washed by water, the aqueous was extracted by CH2Cl2. The comb ined orga nic layer was washed by saturated Na2S2O3 and brine, dried over Na2SO4, and evaporated to give a white solid which was purified by colu mn chromatography eluted with 50% acetic ether in petroleum ether (34 g, 88.6%yield).°C under aRetur nNBS,0.14mol), AIBN (1.32 g)H 2NClClClm-CPBAA solution of 85% m-chloroperoxybe nzoic acid (19 g, 94 mmol) in CH2Cl2 (350 ml)was added at—-0 °C to a solution of 2-Benzylsulfanyl-phenylamine (19 g, 88 mmol) in CH2Cl2 (400 ml). Themixture was allowed to warm to rt in 3 h, then it was washed with a 5% Na2S2O3 soluti on, 10%NaHCO3 solution and brine. The organic layer was dried over Na2SO4, and evaporated. Thesolid was ground in pentane (19 g, 95% yield).Return 氢化反应A mixture of ethyl 3-(N-be nzylam ino )-3-methylbutyrate hydrochloride (25g, 0.1 mol) andlOperce nt Pd-C (2g) in 250 ml of dried alcohol was hydroge nated un der 55 psi H2 for four days.The react ion medium was the n filtered and evaporated un der reduced pressure to provide anamber oil which gradually crystallized upon sta nding (18 g, 100% yield).Retur nHClNH 2ReturnEDCOTo a 0 ° C mixture of BoL-tyrosine (2.04 g, 7.26 mmol) and amylamine (0.63 gl, 7.26 mmol) inmethyle ne chloride (30 ml) is added 1-(3-dimethylami nopropyl)-3-ethylcarbodiimide (EDC) (1.53 resulting solution is diluted with methylene chloride (30 ml) and washed successively with 0.5 M HCl (40 ml), water (20 ml) and sat aq sodium bicarb on ate (25 ml). The orga nic phase is dried over magn esium sulfate and concen trated to a foam (1.84 g, 72.4%yield), sufficie ntly pure to carry into the n ext step. An an alytical sample is obta ined by HPLC.g, 9.9 mmol ). The white mixture is stirred at 0 C for 5 min and at room temp for 23 hrs. TheRetur n三光气成脲To a solution of 2-(tert-butyldimethylsilyloxy)-4-nitroaniline (200 mg, 0.75 mmol) in toluene (10 ml)triethylamine (0.13 ml, 1.64 mmol) and triphosgene (88.4 mg, 0.3 mmol) were added. Thereaction mixture was stirred at 70 ° C for 2 hours, the n cooled to room temperature. Then more 2-(tert -butyldimethylsilyloxy)- 4-n itroa nil ine (200 mg, 0.75 mmol) was added. The result ingmixture was allowed to stir at 70 ° C for 48 hours the n cooled to room temperature. The react ion mixture was partiti oned betwee n water and ethyl acetate. The comb ined orga nic phase waswashed with brine, dried over MgSO4 and filtered. Removal of solve nt at reduced pressure andchromatography of the result ing oil on silica gel (hexa ne: ethyl acetate, 10:1) gave 1,3-Bis-(2-hydroxy-4-nitro-phenyl)-urea (130 mg, 31%yield). Retur n 芳卤用n-BuLi 处理后与Weinreb 酰胺成酮To a solution of diisopropylamine (17.69 ml, 0.135 mole) in THF (200 ml) at argon wasadded n-butyllithium (54.0 ml, 2.5M in hexa ne, 0.135 mole), followed after 5 min bydropwise a solution of 2-fluoro-4-methylpyrid ine (10 g, 0.090 mole) in THF (20 ml). After stirri ng for 15 min at -78 ° C, a solution of Nmethoxy-N-methyl-3-trifluoromethylbenzamide (23.08 g, 0.099 mole) in THF (10 ml) was added dropwise. After stirri ng for more 5 mi n, the react ion wasC and que nched by pouri ng into w^teo ml) and ethyl acetate (400 ml).The layers were separated, and the aqueous layer washed with ethyl acetate (200 ml). The ethyl acetate extracts were comb in ed, dried over an hydrous sodium sulfate, filtered, and concen trated to an oil whichCl O O Cl心丁 3NO 2—78 ° C underallowed to warm to 0was chromatographed on silica gel with 20perce nt ethyl acetate in hexa ne to give 21.6 g of 2-(2-Fluoro-pyridi n-4-yl)-1-(3-trifluoromethyl-phe nyl)-etha none (84.8%yield).Return。
钯催化的插羰反应-060410

药明康德新药开发有限公司
1. 前言…………………………………………………………………2-3 2. 插羰反应制备羧酸及其衍生物…………………………………4-15 3. 插羰反应制备羧酸实验操作……………………………………15-16 4. 插羰反应制备羧酸酯实验操作………………………………16-19 5. 插羰反应制备酰胺实验操作…………………………………19-20 6. 插羰 反应制备 醛…………………………………………………20 7. 插羰反应制备醛实验操作………………………………………21-22 8. 插 羰 反 应 制 备 酮 … … … … … … … … … … … … … … … 22 - 3 0 9. 插羰反应制备酮实验操作………………………………………30-31
HO
+ 2CO + EtOH CHO
PdCl2, Ph3P HCl, 73%
HO MeO
2-24
2-25
ArCHO + HCl
H
Ar
Cl
OH
H
Ar
CO2R
OH
CO 2E t
H
Ar
CO 2 R
Cl
H
Ar
CO2 R
PdCl2(Ph3P)2, Ph3P, BnEt3NCl AcONa, 1 atm., 70oC, 60%
CO2- t-Bu
苄基卤化物的插羰反应还可以在中性的的条件下进行,例如使用分子筛、四甲基尿
素等。在大环内酯弯孢霉菌素 2-20 的全合成中,酯 2-19 的制备就是在这种中性的条件
下通过苄基氯化物的钯催化插羰反应而获得的。
Pd(0) + ArX
CO
O
Ar-P d-X
有机合成 说课稿 教案 教学设计

有机合成 教学目标 1.掌握烃及烃的衍生物性质及官能团相互转化的一些方法;2.了解有机合成的基本过程和基本原则;3.掌握逆向合成法在有机合成中的应用。
教学内容一、课前回顾有机合成的思路就是通过有机反应构建目标化合物的分子骨架,并引入或转化所需的官能团。
你能利用所学的有机反应,列出下列官能团的引入或转化方法吗?1.引入碳碳双键的三种方法是_____________________;_____________________;______________________。
2.引入卤原子的三种方法是__________________________;__________________;_______________________。
3.引入羟基的四种方法是______________;________________;______________;________________。
课前回顾1.卤代烃的消去反应 醇的消去反应 炔烃的不完全加成反应2.烷烃或苯及其同系物的卤代(取代)反应 不饱和烃与HX 、X 2的加成反应 醇与氢卤酸的取代反应3.烯烃与水的加成反应 卤代烃的水解反应(或取代反应) 醛(酮)的加成反应 酯的水解(或取代)反应二、知识点讲解(一)有机合成遵循的原则1.起始原料要廉价、易得、低毒性、低污染。
通常采用四个碳以下的单官能团化合物和单取代苯。
2.应尽量选择步骤最少的合成路线。
为减少合成步骤,应尽量选择与目标化合物结构相似的原料。
步骤越少,最后 产率越高。
3.合成路线要符合“绿色、环保”的要求。
高效的有机合成应最大限度地利用原料分子的每一个原子,使之结合到 目标化合物中,达到零排放。
4.有机合成反应要操作简单、条件温和、能耗低、易于实现。
5.要按一定的反应顺序和规律引入官能团,不能臆造不存在的反应事实。
综合运用有机反应中官能团的衍变规律及 有关的提示信息,掌握正确的思维方法。
有时则要综合运用顺推或逆推的方法导出最佳的合成路线。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
经典化学合成反应标准操作醛酮的经典合成目录1.前言 (4)2.由醇合成醛酮 (4)2.1铬(VI)试剂 (4)2.1.1 Jones氧化(Cr2O3/H2SO4/acetone) (4)2.1.2 Collins氧化(Cr2O3.2Py) (5)2.1.3 PCC(Pyrindium Chlorochromate)氧化 (8)2.1.4 PDC(Pyrindium Dichromate)氧化 (9)2.2 用活性MnO2氧化 (10)2.2.1 用活性MnO2氧化示例一: (10)2.3用DMSO氧化 (11)2.3.1 DMSO-(COCl)2氧化(Swern Oxidation) (11)2.3.2 DMSO-SO3-Pyridine (12)2.4 用氧铵盐氧化 (13)2.4.1 用氧铵盐氧化示例: (13)2.5 用高价碘试剂氧化 (14)2.5 .1 Dess-Martin氧化反应示例: (14)2.5.2 IBX氧化反应示例: (15)2.6 亚硝酸钠和醋酐氧化 (15)2.6.1 亚硝酸钠和醋酐氧化示例 (15)2.6 TPAP-NMO 氧化 (16)2.6.1 TPAP-NMO 氧化示例 (16)2.7 1,2-二醇的氧化 (16)2.7.1 1,2-二醇的氧化示例一: (17)2.7.1 其他1,2-二醇的氧化相关文献: (18)3.由卤化物合成醛酮 (18)3.1 由伯卤甲基和仲卤甲基的氧化合成醛酮 (18)3.1.1 用DMSO氧化(Kornblum反应) (18)3.1.2用硝基化合物氧化(Hass反应) (20)3.1.3用乌洛托品氧化(Sommelet反应) (21)3.1.4用对亚硝基二甲苯胺氧化吡啶翁盐氧化(Kröhnke反应) (22)3.1.5用胺氧化物氧化 (22)3.2 由二卤甲基或二卤亚甲基合成醛酮 (23)3.2.1 由二卤甲基合成醛反应示例: (23)3.3 由有机金属化合物的酰化合成醛酮 (24)3.3.1 由有机金属化合物的酰化合成醛酮示例 (25)3.4 由Pd催化反应合成醛 (25)4.由活泼甲基或活泼亚甲基烷烃合成醛酮 (25)4.1 用SeO2氧化合成醛酮 (26)4.1.1 用SeO2氧化合成醛酮示例 (26)4.2用空气氧化合成酮 (26)4.2.1用空气氧化合成酮反应示例: (27)4.3 用铬酸氧化合成酮 (27)4.3.1 用铬酸氧化合成酮示例 (27)4.4用高锰酸盐氧化合成酮 (29)4.5 用醌氧化合成酮 (29)5.由羧酸及其衍生物合成醛酮 (30)5.1由羧酸合成醛 (30)5.1.1用金属氢化物还原 (30)5.1.2由脱CO2合成醛 (31)5.1.3由羧酸合成酮 (31)5.2由酰氯及酸酐合成醛酮 (33)5.2.1用Rosenmund法合成 (33)5.2.2用金属氢化物还原 (34)5.3由酯及内酯合成醛 (35)5.3.1 酯通过DIBAL还原为醛示例: (36)5.4由酰胺合成醛酮 (36)5.4.1 由酰胺合成醛酮 (37)5.4.2 McFadyen-Stevens Reaction (38)5.5由酯或酰氯经Weinreb酰胺合成醛酮 (39)5.5.1 由Weinreb酰胺还原合成醛反应示例一 (40)5.5.2由Weinreb酰胺还原合成酮反应示例: (41)5.6由氰合成醛酮 (41)5.6.1DIBAL 还原腈到醛示例(最重要的方法) (42)5.6.2Li(EtO)3AlH 还原腈到醛示例(较重要的方法) (43)5.6.3Ranney Ni 加氢还原氰到合成醛示例 (43)5.6.4有机金属试剂对腈加成合成酮示例 (44)6. 由烯烃、芳环合成醛酮 (46)6.1 由烯烃臭氧氧化合成醛 (46)6.2 烯烃用OsO4/NaIO4氧化合成醛 (47)6.3 烯烃经由有机硼化合物中间体的烯烃甲酰化合成醛 (47)6.5 由烯烃的甲酰化合成醛 (48)6.5.1 Vilsmeyer反应 (48)6.5.2 Duff’s 甲酰化 (51)6.5.3 Reimer-Tiemann 甲酰化 (52)6.5.4 Gattermann甲酰化 (53)6.5.5 多聚甲醛/甲醇镁苯酚甲酰化 (53)6.5.6氯化锡/多聚甲醛苯酚甲酰化 (54)6.5.7重氮化后甲酰化 (54)6.6烯烃经加成-氧化反应合成酮 (56)6.6.1 烯烃经加成-氧化反应合成酮示例 (56)7. 由炔烃合成醛酮 (57)7.1 由加成-氧化反应合成醛酮 (57)7.2 由氧化反应合成酮 (57)7.3 由加成-水解反应合成酮 (58)7.4 由加成-还原反应合成酮 (59)7.5 由加成-烷基化,酰化等反应合成酮 (59)8. 由醚及环氧化合物合成醛酮 (59)8.1 Claisen重排 (59)8.2酸催化下环氧化物重排 (61)8.2.1 酸催化下环氧化物重排合成醛酮示例一 (61)8.3氧化法 (61)8.4 水解法缩醛或酮合成醛酮 (61)9. 由胺合成醛 (62)9.1胺的氧化 (62)9.1.1 胺的氧化合成醛反应示例: (63)9.2 由胺经由西佛碱的方法 (64)9.2.1 由胺经由西佛碱合成醛示例 (64)9.3 自苯胺衍生物合成 (64)10. 由硝基化合物合成醛酮 (64)11. 由Friedel-Crafts反应合成芳基酮 (65)11.1 由Friedel-Crafts反应合成芳基酮示例 (68)12. Dieckmann 缩合脱酸 (69)13. 由合成子合成醛酮 (71)14. 由砜合成醛酮 (71)15. Michael 反应和类似反应(Addition, Condensation) (71)1.前言醛和酮是一类重要的有机化合物,其合成在有机合成中占有非常重要的地位。
醛和酮的合成方法繁多,新合成途径也层出不穷。
本部分主要以官能团的转换为主线,依次讨论了由醇、卤化物,甲基、亚甲基、羧酸及其衍生物、烯烃、炔烃、醚及环氧化合物、胺、硝基化合物等转换为醛酮的一些非常实用的方法,一些少用或罕见的反应并没有收录进去。
2.由醇合成醛酮由醇合成醛酮是有机合成中的一类非常重要的反应。
由伯醇的氧化可以得到醛。
由于醛处于醇与羧酸的中间氧化状态,就必须选择适当的氧化剂加以控制,不致氧化过度而生成羧酸。
由仲醇的氧化可以得到酮。
但仲醇过度氧化可以导致分子开裂。
由叔醇的氧化开裂、转位等反应也能合成酮,但实用范围不大。
由此可见,要讨论由醇的氧化就必须从所使用氧化剂氧化性的强弱、醇分子的结构以及反应条件等多个方面入手。
本部分由讨论最常用的铬(VI)氧化剂开始,依次讨论了活性MnO2,DMSO试剂,氧铵盐,高价碘化物等氧化剂在醇氧化合成醛酮反应中的应用。
2.1铬(VI)试剂常用的铬(VI)试剂主要有三氧化铬(CrO3)、重铬酸、铬酸酯[CrO2(OCOR)2]、铬酰氯(CrO2Cl2)等。
为了控制醇不被过度氧化,化学家已经开发了种种氧化方法,最常用的方法有Jones氧化法(Cr2O3/H2SO4/acetone)、Collins氧化法(Cr2O3·2Py)、PCC (Pyrindium Chlorochromate)及PDC(Pyrindium Dichromate)氧化法等。
2.1.1 Jones氧化(Cr2O3/H2SO4/acetone)Jones试剂通常可以将伯醇氧化成酸,把仲醇氧化成酮2.1.1.1(Cr2O3/H2SO4/acetone)合成方法示例A 1-L, round-bottomed flask equipped with a magnetic stirring bar and pressure-equalizing dropping funnel is charged with ethyl 3-hydroxy-4-pentenoate and 400 mL of acetone. The mixture is cooled in an ice bath and Jones reagent (175 mL) is added dropwise via the dropping funnel (addition time is approximately 30–40 min). When addition of the Jones reagent is complete, the reaction mixture is allowed to warm slowly to room temperature and is stirred overnight (10–20 hr). Methanol (20 mL) is added to quench excess Jones reagent and the reaction mixture is poured into a 2-L separatory funnel containing diethyl ether (800 mL). After thorough mixing, the layers are separated and the aqueous layer is extracted with diethyl ether (three 200-mL portions). The combined organic layers are washed with brine (two 200-mL portions), dried over MgSO4, filtered, and the solvent is removed by simple distillation. Final purification is accomplished by Kugelrohr distillation at 0.60 mm (oven temp 45°C) with a 250-mL receiving bulb cooled to −78°C using a dry ice/isopropyl alcohol cold bath. The purified product (14.9 g, 52%) can be stored at −20°C for several months without decomposition.Notes:Jones reagent is prepared by dissolving chromium oxide (CrO3) (23.5 g) in con. sulfuric acid (21 mL) with cooling and then diluting with distilled water to give a total volume of 175 mL.Reference: Organic Syntheses, Coll. Vol. 9, p.432; Vol. 71, p.2362.1.2 Collins氧化(Cr2O3·2Py)Collins氧化法是利用CrO3-pyridine配合物将伯醇和仲醇依次氧化成醛(和/或酸)和酮的方法。