高效液相色谱手性流动相添加剂分离肾上腺素类对映体
高效液相色谱手性固定相法分离酸性化合物对映体

1 36
分析化学
第 32 卷
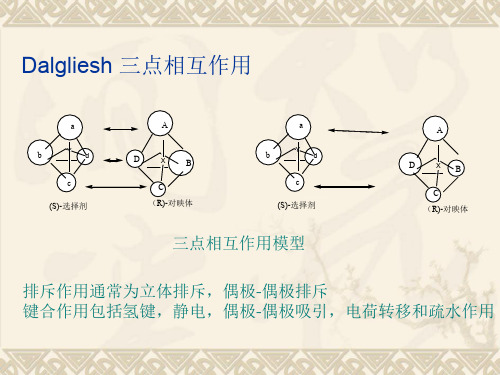
π作用 、氢键作用增强 ; (5) 与 CDMPC 不同 ,环丙烷酸未获得基线分离 。综上所述 ,决定溶质保留和发生手 性识别的关键是溶质与固定相之间的吸引作用 ,主要是氢键作用和π2π作用。
表 2 流动相中异丙醇浓度对酸性手性化合物在 (S ,S)2Whelk2O1 上对映体分离的影响 Table 2 The influence of isopropanol concentration in mobile phase on the enantioseparation of acidic chiral compounds on
第 32 卷
分析化学 (FENXI HUAXUE) 研究报告
第2期
2004 年 2 月
Chinese Journal of Analytical Chemistry
134~138
高效液相色谱手性固定相法分离酸性化合物对映体
蔡小军 徐秀珠 3 张大同 何红梅 潘春秀
3 结果与讨论
3. 1 流动相中异丙醇浓度对手性分离的影响 3. 1. 1 在涂敷型 CDMPC 柱上的分离 改变流动相正己烷中异丙醇的浓度 (含 0. 1 %三氟乙酸) 对这 6 种酸性化合物进行手性分离 ,结果见表 1 。图 1 是它们在 CDMPC 柱上的对映体色谱分离图 。
2003204207 收稿 ;2003208225 接受 本文系浙江省分析测试基金资助项目
1. 10 1. 10 1. 10
1. 59 2. 54 3. 37 1. 38 1. 59 2. 71 0. 03 0. 21 0. 05 1. 25 1. 64 0. 46
1. 23 1. 56 1. 70
k′2. 后洗脱对映体的保留因子 (the retention factor of the second eluted enantiomer) ; 死时间 ( t0) 由 1 ,3 ,52三叔丁基苯测定 (the dead time was determined using 1 ,3 ,52tri2tert2butyl benzene) ;流速 (flow rate) : 0. 5 mL/ min ; 温度 (temperature) : 25 ℃
手性药物对映体的环糊精手性流动相_手性固定相HPLC法拆分

实验 选 择 最 佳 流 动 相 组 成 为 甲 醇 : 0101 mol·L - 1 KH2 PO4 缓 冲 液 ( 25 ∶75 ) , p H 310 。 8021 ,1 ,1’222联萘酚和去甲基安定对映体药物 被拆分的色谱图如图 3 ,分离度列在表 2 。
配合物时 ,并假设 CD 和 CD2溶质的配合物不 被固定相所保留 ,其拆分过程中可能存在如下 平衡式 :
S +A
K
SA
K
=
[ SA ] [ S ][ A ]
S + CD
K1
S CD
K1
=
[ S CD ] [ S ][ CD ]
其中 , S 是被分离的自由溶质 , A 是固定相的
吸附位点 , CD 是环糊精分子 , SCD 是溶质∶环
1. 4 酸度的影响 通常增大酸度 ,有利于碱性 药物的分离 。随 p H 值减小 ,容量因子先是增 大随后又减小 ,而分离度随酸度增加而增大 ,尤 其是 8018 的对映体 ( 1) 随 p H 值减小至 215 时 ,完全达到基线拆分 。 2 β2环糊精浓度的影响及保留机理
根据 Armst rong 报道[2 ] ,β2环糊精作为手 性流动相添加剂进行手性药物对映体拆分时 , 当被拆分的溶质与β2环糊精只形成 1∶1 的包接
·145 ·
均大于 019992 , 可见前面假设在本体系中成 立 ,证实被拆分的溶质与β2环糊精只形成 1∶1 的包接配合物 ,求得的包络常数分别列在表 1 。
食品中L_乳酸检测方法及其研究进展

乳酸作为 1种重要的有机酸广泛存在于生物体中, 是生物 体无氧糖代谢的重要产物。乳酸的分子量为 90. 08, 乳酸分子 含有不对称碳原子, 按其旋光性分为 D 乳酸、L 乳酸、D L乳酸 3种。L 乳酸是 1种较强的有机酸, 能与水互溶, 不易结晶, 当 浓度为 60% 以上时具有很强的吸湿性。L 乳酸可以在食品工 业上用作酸味剂和防腐剂, 与柠檬酸、苹果酸等实用酸相比具 有很强的竞争力 [1]。在美国, L乳酸用于软饮料方面, 在很大 程度上取代了柠檬酸、磷酸等; 在啤酒制造上, 美国禁止使用磷 酸调节 pH 值, 而全部改成 L 乳酸 [2] ; 在肉品加工方面, 猪牛 屠宰后, 胴体 经 乳酸 喷 淋可 有 效 降 低胴 体 上 微生 物 总 数 [ 3- 4] ; 乳酸是肌肉组织内的糖类代谢物质, 动物屠宰后肉 在低温成熟期间肌肉糖原经过无氧酵解生成乳酸, 从而造成 pH 值的下降 [ 5] 。乳酸的堆积量与 pH 值的下降程度呈正相 关 [ 6] 。乳酸大量生成会造成冷却肉保水性及嫩度不同程度 的下降[ 7], 与 PSE 肉、DFD肉的产生有密切关系。
V= A V / t
b。式中 V 为酶促反应速度 ( L
m ol/m in); A 为吸光度变化值; V 为反应液体积 ( m l); t 为时间变化值 (m in); 为摩尔吸收系数 ( L /mmol cm ); b为
高效液相色谱法及其在药物分析中的应用(最新整理)

高效液相色谱法及其在药物分析中的应用以液体为流动相的色谱法称为液相色谱法。
用常压输送流动相的方法为经典液相色谱法,这种色谱法的柱效能低、分离周期长。
高效液相色谱法(highperformanceliquidchromatography,简称HPLC)是在经典液相色谱的基础上发展起来的一种色谱方法。
与经典的液相色谱法相比,高效液相色谱法具有下列主要优点:①应用了颗粒极细(一般为10µm以下)、规则均匀的固定相,传质阻抗小,柱效高,分离效率高;②采用高压输液泵输送流动相,流速快,一般试样的分析需数分钟,复杂试样分析在数十分钟内即可完成;③广泛使用了高灵敏检测器,大大提高了灵敏度。
目前,已经发展了多种不同的固定相,有多种不同的分离模式,使高效液相色谱法的应用范围不断扩大。
下面介绍高效液相色谱法的有关知识,新的方法和技术以及在药物分析中的应用。
一、分类高效液相色谱法按分离机理的不同可分为以下几类:(一)吸附色谱法(adsorptionchromatography)以吸附剂为固定相的色谱方法称为吸附色谱法。
使用最多的吸附色谱固定相是硅胶,流动相一般使用一种或多种有机溶剂的混合溶剂。
在吸附色谱中,不同的组分因和固定相吸附力的不同而被分离。
组分的极性越大、固定相的吸附力越强,则保留时间越长。
流动相的极性越大,洗脱力越强,则组分的保留时间越短。
(二)液-液分配色谱法(liquid-liquidchromatography)液-液分配色谱的固定相和流动相是互不相溶的两种溶剂,分离时,组分溶入两相,不同的组分因分配系数(K)的不同而被分离。
目前广泛使用的化学键合固定相是将固定液的官能团键合在载体上而制成的,使用化学键合固定相的色谱方法(简称键合相色谱法)可以用分配色谱的原理加以解释。
键合相色谱法在HPLC中占有极其重要的地位,是应用最广的色谱法。
按照固定相和流动相极性的不同,分配色谱法又可分为正相色谱法和反相色谱法两类。
手性药物的拆分——高效液相色谱(HPLC)
其它类型的旋光性聚合物CSPs还包含旋光性聚酰 胺、聚氨酯类、含有轴手性联萘基的旋光性聚合物、 (+)-聚-N-二苯甲基马来酰亚胺等。
2.手性拆分实例
2.1 高效液相色谱手性固定相法分离酸性化合物对 映体
手性柱 涂敷型CDMPC(20 nm, 300 mm×4.0 mm) Pirkl(S,S)WhelkO1(12mm,250mm×4.0mm )
1.4 多模式环糊精固定相
环糊精衍生化固定相既可以用于反相分离又可以用于正相分 离,因而称为多模式手性固定相。手性识别作用既有包容络 合,又有π-π电荷转移,氢键和立体排斥相互作用等多种作 用,应用范围广泛。
制备过程有四个步骤:
(1)硅胶上键合N-(2-氨乙基-3-氨丙基)基团 (2)用对甲苯磺酰氯专一性的磺化环糊精的羟基 ( 3)2-氨乙基-3-氨丙基键合硅胶与甲苯磺酰氯-β-环糊精反应;通
淀粉手性固定相的制备方法与纤维素手性固定相相似,首先 用淀粉与相应的有机化合物,如苯甲酰氯,苯基异氰酸酯等 反应,得到淀粉衍生物,再将这种衍生物涂敷在氨丙基键合 硅胶上,便可以得到淀粉类手性固定相。
1.3 环糊精手性固定相
OH
HO O OHOHO
OHO OH O
HO
OH
OOH
O
OH
O
HO
OH
+
SiO2
O O Si O
CH2CH2CH2NHCOCH2CH2CONHCH2CH2NH
3.碳氧键连接 醚基键合相与环糊精反应,:
O
SiO2
O
HO
O Si
+ (CH2)3oCH2CHCH2
O
SiO2
O O Si O
分子印迹手性拆分详解

例:反相 HPLC 法, 直接进样生物样品来评价新型限进 CSP—poly-CD-RAM, 结果显示, 样品中的蛋白质先被洗脱, 对检测无干扰,同时将其直接注入人血 浆和牛奶中其中进行HPLC分析可以得到的几种手性药物和农药均能够在 poly- CD-RAM 上实现手性拆分。
五、高效液相色谱手性流动相添加剂法分离手性药物 1、定义:在流动相中添加手性选择剂, 制造手性环境使外消旋 体分离,即手性流动相添加剂(chiral mobile phase additive, CMPA)法。对映体和手性选择剂相互作用形成瞬态非对映体复合 物,两复合物稳定性及其在固定相和流动相之间分配行为的不同 使其在非手性色谱柱从而实现分离的目的。 2、优缺点;CMPA 法拆分对映体不需要柱前衍生化,常规色谱 柱即可分离,成本相对较低且更加灵活,针对不同样品可使用不 同手性添加剂,但是某些手性流动相添加剂不稳定,干扰检测结 果。 3、例:以 β- 环糊精作为手性流动相添加剂,研究佐匹克隆对映体在反相高效
以拆分手性药物酮洛芬为例:采用 4-乙烯基吡啶为功能单体,以二甲 基丙烯酸乙二醇酯为交联剂,引
发剂偶氮二异丁腈(AIBN).在模板
分子(S)-酮洛芬的存在下,制备出 (S)-酮洛芬的分子印迹聚合(MIPs), 并用匀浆法装柱 (250mm×4mm,i.d.).
然后使用V(乙酸)∶V(乙腈)=
1∶9 的溶液洗脱,至无模板分子, 用HPLC进行分析. 色谱条件:流动相为乙腈(含有一定 量乙酸);UV检测,波长258 nm;流
速0.2 mL/min;进样体积10μL;操作
温度19~36℃ 。
(R)-和(S)-酮洛芬在印迹柱上的分离谱图
(S)-酮洛芬在印迹柱上的谱图
空白柱没有拆分能力,而制得的分子印迹聚 合物对酮洛芬的外消旋混合物显示出良好 的手性拆分能力,(S)-酮洛芬的容量因子为
固相萃取-高效液相色谱手性流动相添加剂法测定血浆中氧氟沙星对映体

固相萃取-高效液相色谱手性流动相添加剂法测定血浆中氧氟沙星对映体王乃毅;白小红【期刊名称】《山西职工医学院学报》【年(卷),期】2012(022)001【摘要】目的:建立固相萃取-高效液相色谱法(high performance liquid chromatogoraphy,HPLC)拆分及测定血浆中氧氟沙星对映体的方法.方法:以L-异亮氨酸为配合剂,Cu2+为配合离子,采用HPLC手性流动相添加剂法测定血中氧氟沙星对映体的含量.色谱柱为ODS-BP柱(250 mm×4.6 mm,5μm),内标物为环丙沙星,流动相为手性添加剂溶液(含1.31 g/L L-异亮氨酸和0.80 g/L CuSO4)-甲醇(81.5∶18.5),激发波长为330 nm,发射波长为504 nm,流速为0.8 mL/min.其中,应用C18固相萃取小柱萃取血浆中的氧氟沙星.结果:在经过优选的色谱条件下,对映体可以良好分离,分离度达到1.5以上;S-(-)-与R-(+)-氧氟沙星的线性范围均为0.08~20 μg/mL(r分别为0.994 5和0.994 7),最小检测限均为4 ng/mL,平均萃取回收率分别为89.77%和88.07%,平均回收率分别为97.19%和98.39%,日内RSD分别为2.9%和3.0%,日间RSD为2.0%和2.2%.结论:该法血样处理简单,干扰小,灵敏度高,精密度好,可以作为对血浆中左、右旋氧氟沙星分别测定的方法.【总页数】4页(P1-4)【作者】王乃毅;白小红【作者单位】山西职工医学院,山西太原030012;山西医科大学,山西太原030001【正文语种】中文【中图分类】R927.2【相关文献】1.高效液相色谱法分离药物对映体中手性流动相添加剂的应用 [J], 祝艳琳;章靖;田丹碧2.高效液相色谱手性流动相法测定盐酸肾上腺素注射液中 S-对映体含量 [J], 金丽;周建丽;张晓丹;古卓良;卜莹3.正相高效液相色谱-手性配位基交换流动相法测定人血清中的甲状腺素对映体 [J], 王荣;谢景文;贾正平;胡晓丽;谢华;李永民;陈立仁4.RP-HPLC手性流动相添加剂法分析尿中氧氟沙星对映体 [J], 曾苏;章立;刘志强5.高效液相色谱手性流动相添加剂法拆分愈创甘油醚对映体 [J], 翟明翚;韩爽;王颖;陈志伟;苏立强因版权原因,仅展示原文概要,查看原文内容请购买。
手性药物分离CRD、CMP和CSP方法的特点

试述手性药物分离CRD、CMP和CSP方法的特点。
HPLC用于手性分离概括起来可分为两大途径:间接法和直接法。
其中直接法包括手性流动相添加剂法(CMP)和手性固定相法(CSP),间接法(CDR)又称为非对映体拆分法或柱前手性衍生化法。
间接法(CDR),是手性化合物对映体在分离之前把手性药物对映体混合物在预处理液中进行柱前衍生化,形成一对非对映异构体,非对映体对与固定相之间的键合力如偶极-偶极、电荷转移、氢键、疏水性不等,产生差速迁移而被分离。
其优点在于:可采用通用的非手性柱分离;通过衍生化可提高检测灵敏度;分离条件简单;分离效果好。
而其缺点在于:要有可被衍生化的基团;要有高光学纯度的手性试剂;对个对映体衍生化率速和平衡常数应一致;衍生化和色谱过程中不能发生消旋化。
手性流动相添加剂法(CMP)是将手性试剂加到LC流动相中,与手性药物生成可逆的非对映体复合物,根据复合物的稳定性,在流动相中的溶解性和与固定相的键合力差异,于非手性固定相上分离对映体。
优点是:不需昂贵的手性柱,亦无须进行柱前衍生,手性添加剂可视要求而更换,可变范围较宽,使用比较方便。
缺点是:可拆分的化合物范围有限;某些添加剂不够稳定且往往会干扰测验。
手性固定相法(CSP)是将手性试剂化学键合到固定相上制成手性柱,外消旋体中的一个手性物质与手性固定相发生作用,生成不稳定的短暂复合物,而两者在固定相中的保留时间不同,从而达到分离的目的。
优点是:能广泛适用于各类化合物,适于常规及生物样品的分析测定;除非必须衍生化,否则无需高光学纯度试剂;样品处理步骤简单;制备分离方便,定量分析的可靠性较高;缺点是:样品有时也须作柱前衍生(但不一定是手性衍生化试剂);对样品结构有一定限制,其适用性尚不及普通HPLC固定相(包括正相和反相)那样广泛。
该方法是目前认为非常简单有效,而且应用最为广泛的方法,迄今为止,CSP柱商品已有40多种,但价格大多昂贵,尚未有一种具有类似ODS柱的普遍适用性。