动物组织中阿莫西林残留的液相色谱分析方法研究
高效液相色谱串联质谱法测定动物组织中林可酰胺类和大环内酯类抗生素残留

高效液相色谱串联质谱法测定动物组织中林可酰胺类和大环内酯类抗生素残留岳振峰;陈小霞;谢丽琪;吉彩霓;华红慧【期刊名称】《分析化学》【年(卷),期】2007(35)9【摘要】建立了动物组织样品中洁霉素、氯洁霉素、红霉素、螺旋霉素、交沙霉素、泰乐菌素、竹桃霉素等7种林可酰胺类及大环内酯类药物多残留的液/质联用确证方法.用乙腈萃取样品中7种林可酰胺类及大环内酯类抗生素,然后用正己烷脱脂,旋转蒸发仪浓缩,以Luna C18(2)色谱柱为分离柱,在正离子模式下以电喷雾电离串联质谱仪进行测定.方法的检出限为0.05~5.3 μg/kg.在20 、50、200 μg/kg 3个浓度水平进行验证实验,方法的线性范围为20~200 μg/kg;总体平均回收率为70.3%~102%;相对标准偏差为2.4%~16.6%.本方法简便、快速、准确,各项技术指标满足国内外法规的要求,可用于动物组织样品中林可酰胺类及大环内酯类抗生素残留的确证检测.【总页数】5页(P1290-1294)【作者】岳振峰;陈小霞;谢丽琪;吉彩霓;华红慧【作者单位】深圳出入境检验检疫局食品检验检疫技术中心,深圳,518067;深圳大学教务处,深圳,518060;深圳出入境检验检疫局食品检验检疫技术中心,深圳,518067;深圳出入境检验检疫局食品检验检疫技术中心,深圳,518067;深圳出入境检验检疫局食品检验检疫技术中心,深圳,518067【正文语种】中文【中图分类】O6【相关文献】1.超高效液相色谱串联质谱法测定鳗鱼中大环内酯类和林可酰胺类抗生素残留量的研究 [J], 刘正才;杨方;林永辉;张琼;刘素珍;苏芝娇;潘迎芬2.液相色谱串联质谱法测定动物组织中大环内酯类残留量 [J], 徐娜;王亚立;王爱芹;任召珍;刘志敏3.超高效液相色谱串联质谱法对鳗鱼中大环内酯类、喹诺酮类和磺胺类兽药残留量的同时测定 [J], 陈莹;陈辉;林谷园;林荆;江滨炜;吴文凡4.高效液相色谱-串联质谱法测定河蟹肝胰腺组织中林可酰胺类抗生素残留 [J], 邱稀木;朱晓华;边文冀;王淑芳;孟勇5.高效液相色谱串联质谱法测定牛奶中林可酰胺类和大环内酯类抗生素残留量的研究 [J], 谢丽琪;岳振峰;唐少冰;陈小霞;吉彩霓;华红慧因版权原因,仅展示原文概要,查看原文内容请购买。
HPLC法测定阿莫西林克拉维酸钾颗粒的含量

HPLC法测定阿莫西林克拉维酸钾颗粒的含量【摘要】目的:使用高效液相色谱法对阿莫西林克拉维酸钾颗粒进行检测,测定其成分组成的含量。方法:采用PhenomenexC18(2)色谱柱(4.6mm15mm,5μm),岛津LC-10A高效液相色谱仪,以0.78%的磷酸二氢钠水溶液(pH值为4.4)一甲醇(95:5)为流动相,流速0.8mL/min,柱温30℃,检测波长220nm,进样体积为10μL,分析时间为10min,外标法定量。结果:阿莫西林在质量浓度为0.60~1.20mg/mL浓度范围内线性关系良好,相关系数为0.9999,平均回收率(n=9)为99.32%,RSD 为O.29%。克拉维酸在O.10~0.30mg/mL浓度范围内线性关系良好,相关系数为O.9999,平均回收率(n=9)为99.08%,RSD为0.28%。克拉维酸钾与阿莫西林标示量分别为90.0%一110.0%。结论:此种测定方法操作较为简便,且测量效果好,准确灵敏度高,在质量测定工作中可大范围使用。【关键词】阿莫西林;克拉维酸钾;HPLC含量测定作为阿莫西林与克拉维酸钾的复方制剂,阿莫西林克拉维酸钾的体内外抗菌活性与临床试验研究发现克拉维酸钾能提高阿莫西林的药品可以有效对耐药菌活性进行抑制,可以获得更为稳定且确切的疗效,同时对革兰阳性以及革兰阴性细菌均可产生疗效。本文采用HPLC法对阿莫西林克拉维酸钾颗粒的含量进行测定,并在色谱柱、流速以及样品选择上进行了改进,让分离度、理论塔板数和拖尾因子最终结果均优于USP标准,可以获得较好的结果,且操作方便。1仪器与试剂高效液相色谱仪:岛津LC-10A高效液相色谱仪;SPD-6V紫外检测器;HS色谱工作站;电子天平(赛多利斯BP211D); Millipore超纯水机。阿莫西林和克拉维酸钾对照品(中国药品生物制品检定所提供);甲醇为色谱纯(美国TEDLA公司),磷酸二氢钠氢氧化钠均为分析纯(国药集团化学试剂有限公司);试验用水均为超纯水。阿莫西林克拉维酸钾颗粒(由先声药业公司提供,产品批号为20060403、20060404、20060405)。2方法与结果2.1色谱条件及系统适用性试验色谱柱:PhenomenexC18(2)柱(4.6mmx150mm,5pm);流动相:0.78%的磷酸二氢钠缓冲液[用磷酸或1mol/L的氢氧化钠调pH值至(4.4+0.1)]-甲醇(95:5);检测波长为220nm;流速为0.8mU/min;柱温30C;进样体积10μL;理论塔板数按阿莫西林峰和克拉维酸峰分别计算,应均不低于2000。阿莫西林和克拉维酸之间的分离度应不小于3.5。2.2供试品溶液与对照品溶液的制备供试品溶液的制备:取阿莫西林克拉维酸钾颗粒10包,混合均匀,精密称取适量,用流动相溶解稀释并过滤。制成每毫升含克拉维酸和阿莫西林分别为0.25mg和1.0mg的溶液。对照品溶液的制备:精密称取克拉维酸钾对照品和阿莫西林对照品适量,用流动相溶解过滤、稀释,制成每毫升含克拉维酸和阿莫西林分别为0.25mg和1.0mg的溶液。2.3测定方法精密量取对照品及供试品溶液各10μL,分别注入高效液相色谱仪,记录色谱图,量取峰面积,按外标法计算供试品中阿莫西林(C16H19N3O5S)及克拉维酸(C8H9NO5)的含量,即得。2.4标准曲线2.4.1精密称取阿莫西林对照品150.08mg,置50mL量瓶中,加流动相溶解并稀释至刻度,精密量取上述溶液2.0、2.5、3.0、3.5和4.0mL,分别置10mL量瓶中,加流动相稀释至刻度,摇匀,分别精密量取10μL注入液相色谱仪,结果见表1。由表中数据进行线性回归求得直线方程:A=21646692C+46800.4,r=0.9999表明阿莫西林在0.60~1.20mg/mL浓度范围内线性关系良好。2.4.2精密称取克拉维酸钾对照品50.09mg,置50mL量瓶中,加流动相溶解并稀释至刻度,精密量取上述溶液1.0、1.5、2.0、2.5、3.0mL。分别置10mL量瓶中,加流动相稀释至刻度,摇匀,分别精密量取10注入液相色谱仪,结果见表2。由表中数据进行线性回归求得直线方程:A=4905430C—122744,r=0.9999表明克拉维酸钾在0.10~0.30mg/mL浓度范围内线性关系良好。2.5最小检出限和定量限2.5.1取阿莫西林对照品适量,加流动相溶解配制成浓度为1.0mg/mL的溶液,然后逐步稀释至原浓度的1/100、1/200、11300、11400、11500,按上述色谱条件进样测定,最小检出限为2μg/mL (SIN=3),最小定量限为10μg/mL (SIN=10)。2.5.2取克拉维酸钾对照品适量,加流动相溶解配制成浓度为0.5mg/mL的溶液,然后逐步稀释至原浓度的1/100、11200、11300、1/400、1/500按上述色谱条件进样测定,测得最小检出限为1μg/mL (SIN=3),最小定量限为5μg/mL (S/N=lO)。2.6精密度试验取含量测定项下同一样品液(批号:20060403),按上述色谱条件重复进样6次进样测定,以相应的色谱峰的面积计算RSD,阿莫西林为0.15%、克拉维酸为0.06%(n=6),本方法测定阿莫西林和克拉维酸钾有较好的重现性。2.7加样回收率试验按处方比例称取辅料制成的空白颗粒,分成9等份,3份为一组,分别精密加入一定量的阿莫西林对照品和克拉维酸钾对照品,充分混合均匀,置于50mL量瓶中,并按含量测定方法进行操作。2.8稳定性试验按照含量测定项规定进行对照品溶液制备,制成每毫升含克拉维酸与阿莫西林质量分数为O.25和1.0mg的溶液,并严格按照测定方法上的操作步骤规定进行实验,在1、4、6、的时间段上进样。
阿莫西林
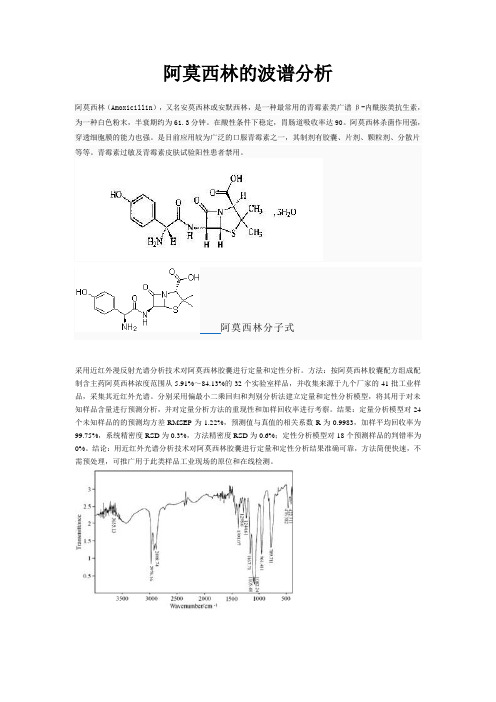
阿莫西林的波谱分析阿莫西林(Amoxicillin),又名安莫西林或安默西林,是一种最常用的青霉素类广谱β-内酰胺类抗生素,为一种白色粉末,半衰期约为61.3分钟。
在酸性条件下稳定,胃肠道吸收率达90。
阿莫西林杀菌作用强,穿透细胞膜的能力也强。
是目前应用较为广泛的口服青霉素之一,其制剂有胶囊、片剂、颗粒剂、分散片等等。
青霉素过敏及青霉素皮肤试验阳性患者禁用。
阿莫西林分子式采用近红外漫反射光谱分析技术对阿莫西林胶囊进行定量和定性分析。
方法:按阿莫西林胶囊配方组成配制含主药阿莫西林浓度范围从5.91%~84.13%的32个实验室样品,并收集来源于九个厂家的41批工业样品,采集其近红外光谱。
分别采用偏最小二乘回归和判别分析法建立定量和定性分析模型,将其用于对未知样品含量进行预测分析,并对定量分析方法的重现性和加样回收率进行考察。
结果:定量分析模型对24个未知样品的的预测均方差RMSEP为1.22%,预测值与真值的相关系数R为0.9983,加样平均回收率为99.75%,系统精密度RSD为0.3%,方法精密度RSD为0.6%;定性分析模型对18个预测样品的判错率为0%。
结论:用近红外光谱分析技术对阿莫西林胶囊进行定量和定性分析结果准确可靠,方法简便快速,不需预处理,可推广用于此类样品工业现场的原位和在线检测。
阿莫西林药物分析:方法名称:阿莫西林原料药-阿莫西林-高效液相色谱法应用范围:本方法采用高效液相色谱法测定阿莫西林原料药中阿莫西林的含量。
本方法适用于阿莫西林原料药。
方法原理:供试品经流动相溶解并定量稀释,进入高效液相色谱仪进行色谱分离,用紫外吸收检测器,于波长254nm处检测阿莫西林的峰面积,计算出其含量。
试剂: 1. 乙腈2. 磷酸二氢钾溶(0.05mol/L)3. 氢氧化钾溶液(2mol/L)仪器设备: 1. 仪器1.1 高效液相色谱仪1.2 色谱柱十八烷基硅烷键合硅胶为填充剂,理论塔板数按阿莫西林峰计算应不低于2000。
高效液相色谱法分析牛和猪肌肉组织中残留的安乃近药物的三种代谢物

收稿日期: !""# +"( +$* 通讯联系人: 沈金灿, 男, 博士, ,$) : ( "#%% ) !&&’"&#" , -+%./) : 012 + 032/4* 5"#* 26* 基金项目: 国家标准委员会计划资助项目 ( !"* !""%$’%, +,+*!& ) *
经首 过 作 用 后, 几 乎 全 部 转 变 成 988 后 再 进 入 体 循环, 其药理作用主要由 988 产生, 988 在体内代 谢为 ( + 甲酰氨 基 安 替 比 林 ( 788 ) 和 (+氨 基 安 替 比
表 ! " 安乃近代谢物测定的梯度洗脱条件 :*6+’ ! " ;.*2"’5- ’+/-"05 10. -&’ 2’-’.("5*-"05 01 2"34.05’ (’-*60+"-’, ! " 8,/ $ #( #+ #* %& ( G90.2 ) "H ! /$ &$ ( /$ /$ ( $.0I9/;- ) "H ! #$ +$ /( #$ #$
阿莫西林制剂的含量测定技术

阿莫西林制剂的含量测定技术【摘要】药物阿莫西林的原有剂型有片剂、胶囊剂等,目前又出现了一些新的剂型、制剂,为了适应新剂型、制剂的要求和进一步完善阿莫西林原有的质控标准,文章对测定阿莫西林制剂含量的方法进行了研究,并在本文中作了阐述。
【关键词】阿莫西林;测定技术作为光谱抗生素药物的一种,临床上阿莫西林被广泛应用,其又名为羟氨苄青霉素,属β-内酰胺青霉素类抗生素。
作为阿莫西林研究的一个重要方面,阿莫西林制剂的含量的测定方法一直备受关注。
文章对测定阿莫西林制剂含量的方法进行了研究。
测定阿莫西林制剂含量的测定方法主要有微生物效价测定法、碘量法、电位滴定法、旋光法、分光光度法以及流动注射化学发光法等,下面对以上这几种方法一一说明。
1.微生物效价测定法作为抗生素测定经典方法之一,微生物效价测定法同样适用于阿莫西林制剂含量的测定,通过该种方法测定出的数据能够直接反应阿莫西林对病菌微生物的抑杀能力,但该种方法的不足之处是实验操作繁琐、工作量较大。
具体方法是以藤黄八叠球菌(28001)为试验菌种,试验培养基为药典Ⅰ号培养基进行实验,最低检测浓度为0.05mg/L,日内和日间变异系数小于15%,线性范围为0.05~400mg/L(r=0.9977,P<0.01),符合生物样品分析要求。
2.碘量法碘量法是利用阿莫西林分子不消耗碘,而其降解产物消耗碘的特性来测定阿莫西林含量的方法,也是一种测定青霉素类抗生素较为通用的方法。
具体方法是先将阿莫西林制剂水解成阿莫西林噻唑酸,然后用足够的、定量的碘与其发生反应,之后再用准硫代硫酸钠滴定剩余的碘,从而测出消耗碘的阿莫西林的量,最终测出制剂中阿莫西林的含量。
3.电位滴定法电位滴定法是利用阿莫西林的降解产物能够与汞盐(Hg2+)发生反应,形成巯基化物汞盐这一特性而得以测定制剂中阿莫西林含量的,青霉素类化合物的降价降解产物普遍存在这一特性。
该种方法先将阿莫西林制剂水解,然后以铂电极为指示电极、Hg-Hg2SO4为参比电极,然后用硝酸汞滴定液(0.02mol/L)滴定,最终测得制剂中阿莫西林的含量。
液相色谱-串联质谱法快速测定水生动物药材中硝基呋喃类代谢物残留量

液相色谱-串联质谱法快速测定水生动物药材中硝基呋喃类代谢物残留量发布时间:2021-08-17T08:28:29.380Z 来源:《科技新时代》2021年5期作者:张朝云[导读] 随着我国中药现代化项目的实施,中药材的安全性越来越受到关注。
养生堂药业有限公司 570216摘要:建立水生动物药材样品中4 种硝基呋喃类代谢物残留快速检测的高效液相色谱-串联质谱法。
方法:硝基呋喃类代谢物经过二硝基苯甲醛衍生化(超声波辅助),用乙酸乙酯提取,经液相色谱分离,串联四极杆质谱多反应监测(MRM) 模式检测,内标法定量。
结果: 4种硝基呋喃类代谢物线性范围为0.0~20.0 ng/mL,相关系数均大于0.99,线性范围良好,方法检出限为0.5μg/kg,在不同质量浓度的添加水平下,4种代谢物的平均回收率在94.3%~115.3%之间,相对标准偏差<10%。
试验对比了快速衍生和恒温振荡衍生的效果,两种方法所测得的含量偏差<10%,本方法相较于恒温振荡衍生法更为简便、快速,显著缩短样品前处理时间,且灵敏度和准确度较高,适合对水生动物药材中硝基呋喃类代谢物残留量的检测。
关键词:高效液相色谱-串联质谱法(LC-MS-MS);超声辅助衍生;水生动物药材;硝基呋喃类代谢物。
引言随着我国中药现代化项目的实施,中药材的安全性越来越受到关注。
硝基呋喃类药物作为广谱抗菌药,曾在养殖业中被广泛应用于预防和治疗由沙门氏菌和埃希氏菌引起的胃肠道疾病。
但后来的研究发现,硝基呋喃类药物及其代谢物具有遗传毒性和致癌性,我国农业部于2002年颁布禁止使用硝基呋喃类抗生素的禁令。
但在实际生产实践中,该类药物以抗菌效果好、抗菌谱宽、价格低等特点,使得该类药物在畜禽养殖业和水产养殖业中仍然存在违法使用的可能性,给人类健康造成威胁。
硝基呋喃类药物进入动物体内后迅速代谢,无法直接检测动物组织中的原型药物,药物代谢后形成相应的代谢产物,分别为AMOZ、AOZ、SEM、AHD,代谢产物与动物机体蛋白质紧密结合形成稳定的化合物,因此,常以检测其代谢物作为残留标志物。
利用高效液相色谱法 高效检测动物源性食品中β-内酰胺类抗生素残留
利用高效液相色谱法高效检测动物源性食品中β-内酰胺类抗生素残留作者:刘彦钊张丽丽来源:《中国食品》2020年第18期近些年来,由于β-内酰胺类抗生素细菌耐药性通过食物链从动物源性食品向人体的转移,消费者越来越担心畜牧业及水产养殖业中的抗生素滥用情况。
动物源性食品中β-内酰胺类抗生素的低浓度水平和基质复杂性,使得在测定其残留量时必须使用高度敏感、高特异性的测定方法。
目前,高效液相色谱法(HPLC)联合其他检测仪器已被广泛用于动物源性食品中β-内酰胺类抗生素残留的检测分析。
一、高效液相色谱-紫外检测法目前,通过使用二极管阵列检测器(DAD)进行UV检测是与HPLC结合使用的非常流行的技术,但在确定β-内酰胺方面存在一些局限性,例如,由于缺乏生色团而导致灵敏度较低,HPLC-UV甚至在不使用DAD的情况下也不能完全可靠地进行β-内酰胺确认。
二、高效液相色谱-串联质谱法质谱法是确定食品中是否存在β-内酰胺的首选检测技术。
尽管此检测技术具有很高的选择性,但仍需要先进行色谱分离,以避免共洗脱化合物的离子抑制。
β-内酰胺首先使用热喷雾通过HPLC-MS测定,由于β-内酰胺电离的简便性,目前电喷雾电离(ESI)是这两种技术之间的首选接口。
尽管β-内酰胺具有羧基,但是由于灵敏度较高,通常使用正离子化。
三、高效液相色谱-化学发光法由于其固有的灵敏度和选择性,化學发光检测是抗生素检测中常用的方法。
β-内酰胺可以通过两种方式参与化学发光反应:增强化学发光发射;用合适的化学发光试剂衍生化。
目前与高效液相色谱联用检测β-内酰胺抗生素的化学发光体系常见的有KMnO4、Ce (IV)、鲁米诺、Ru(bpy)3 2+等。
四、高效液相色谱-荧光分析法目前已有使用高效液相色谱法联合荧光检测器测定β-内酰胺的相关报道。
由于β-内酰胺类化合物中缺少荧光团,通常需要衍生化,而衍生化造成处理过程繁琐,所以效率不佳。
一般来讲,使用化学发光能够检测的β-内酰胺化合物也可使用荧光分析检测,但化学发光可获得更好的灵敏度。
超高效液相色谱_串联质谱法测定动物源食品中7种药物残留
超⾼效液相⾊谱_串联质谱法测定动物源⾷品中7种药物残留超⾼效液相⾊谱-串联质谱法测定动物源⾷品中7种药物残留张崇威,班付国,宋志超,陈蔷,吴宁鹏(河南省兽药监察所,郑州450008)[收稿⽇期]2014-07-01⼀[⽂献标识码]A⼀[⽂章编号]1002-1280(2014)08-0051-04⼀[中图分类号]S859.84[摘⼀要]⼀建⽴了猪⾁⼆鸡⾁⼆鸡蛋中阿奇霉素⼆克林霉素⼆罗红霉素⼆头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定7种药物残留的液相⾊谱-串联质谱法三采⽤⼄腈?⽔=15?2的溶液作为提取液,经正⼰烷净化,UPLC-MS/MS测定三结果表明:以上七种药物在2100µg/L范围内线性关系较好,相关系数r均在0.998以上三在猪⾁⼆鸡⾁和鸡蛋组织中阿奇霉素⼆克林霉素⼆罗红霉素的检测限为0.5µg/kg,定量限为1µg/kg,头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定检测限为2µg/kg,定量限为4µg/kg三在4⼆8⼆20µg/kg的添加浓度上进⾏回收率试验,阿奇霉素⼆克林霉素⼆罗红霉素回收率在80%100%之间,头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定回收率在70%90%之间三本⽅法批内相对标准偏差?20%,批间相对标准偏差?20%三[关键词]⼀头孢类;⼤环内酯类;残留;液质联⽤作者简介:张崇威,硕⼠,从事畜产品兽药残留分析三E-mail:zhang.wei4@163.comDeterminationofSevenDrugsinAnimalOriginFoodbyUltraPerformanceLiquidChromatography-TandemMassSpectrometryZHANGChong-wei,BANFu-guo,SONGZhi-chao,CHENQiang,WUNing-peng(HenanInstituteofVeterinaryDrugControl,Zhengzhou450008,China)Abstract:Amethodbasedonulraperformanceliquidchromatographycoupledwithtandemmassspectrometry(UPLC-MS/MS)wasdevelopedfordeterminationofsevendrugsinpork,chickenandegg.Theanalyteswereextractedwithacetonitrile-water(V/V,15?2),andwerecleanedupbyhexane.QuantificationoftheanalyteswasachievedbyUPLC-MS/MSwithmultiplereactionmonitoring(MRM)usingexternalstandardmethod.Theresultsindicatedthatthecalibrationcurvesweregoodlinearbetweenthepeakareasandconcentrationsof7drugsintherangeof2100µg/L,withcorrelationcoefficientsmorethan0.998.Thelimitsofdetectionandlimitsofquantificationofazithromycin,clindamycin,roxithromycininpork,chickenandeggtissuewere0.5µg/kgand1µg/kg,thelimitsofdetectionandlimitsofquantificationofcefalexin,cephapiriin,cefotaxime,cefradineinpork,chickenandeggtissuewere2µg/kgand4µg/kg,respectively.Theaveragerecoveriesofazithromycin,clindamycin,roxithromycinatthreeconcentrationlevelsof4µg/kg,8µg/kgand20µg/kgrangedfrom80%to100%,andtheaveragerecoveriesofcefalexin,cephapiriin,cefotaxime,cefradinerangedrangedfrom70%to90%.Theintra-andinter-RSDswerelessthan20%.Keywords:cephalosporin;macrolide;residues;UPLC-MS/MS⼀⼀随着畜牧业发展,⽤于预防和治疗禽畜疾病的兽药品种也不断增加,⼈药兽⽤以及兽⽤药物残留超标的现象也越来越多[1]三阿奇霉素⼆克林霉素⼆罗红霉素⼆头孢匹林⼆头孢拉定属于⼈⽤药物,尚未批准在兽医临床上使⽤,但在实际的畜产品药物残留检验中却时有发现三头孢氨苄⼆头孢噻肟作为常⽤兽药在养殖环节中被滥⽤导致残留量超标现象严重三造成这种现象的原因,主要是由于⼀些不法养殖者在养殖过程中违规使⽤,从⽽造成在禽畜产品中的残留超标三为打击和防范违法使⽤和滥⽤这些药物,有必要建⽴这7种药物残留的同步检测技术三⽬前,对于此7种药物的单个或多个检测⽅法也分别有报道,已报道的有13种林可胺类及⼤环内酯类药物的检测[2],以及13种β-内酰胺类药物残留检测[3],但是同时测定该7种药物残留的检测⽅法尚未见报道三本研究建⽴了7种药物残留的快速⼆准确⼆同步检测的液相⾊谱-串联质谱⽅法三1⼀材料与⽅法1.1⼀仪器⼀AcquitUPLC-XevoTQ-S质谱联⽤仪(Waters公司);3K-30台式⾼速冷冻离⼼机(Sigma公司);R215Professional旋转蒸发仪(瑞⼠Buchi公司);IKAMS3.Basic圆周振荡器(⼴州仪科实验室技术有限公司);ALC-2100.2电⼦天平(赛多利斯科学仪器(北京)有限公司);VX-Ⅲ多管涡旋振荡器(北京踏锦科技有限公司)三1.2⼀试剂⼀甲醇⼆⼄腈⼆甲酸均为⾊谱纯,实验⽤⽔为⼀级⽔;阿奇霉素⼆克林霉素⼆罗红霉素⼆头孢氨苄标准品来源于中国兽医药品监察所,头孢匹林⼆头孢噻肟⼆头孢拉定标准品来源于中国药品⽣物制品检定所三1.3⼀标准贮备液的配制⼀分别精密称取7种标准品各约10mg,置于10mL量瓶中,阿奇霉素⼆克林霉素⼆罗红霉素标准品⽤⼄腈溶解并稀释成约1mg/mL的标准贮备液;头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定⽤50%⼄腈溶液溶解,并稀释成浓度约1mg/mL的标准贮备液三然后⽤50%⼄腈溶液逐步稀释成适当浓度的混合标准⼯作液三1.4⼀⽅法1.4.1⼀⾊谱条件⼀⾊谱柱:WatersAcquityUPLCTMBEHC18(2.1mm?50mm,1.7µm);柱温30?;流动相A:⼄腈,B:0.1%甲酸溶液;进样量5µL;梯度洗脱程序见表1三表1⼀⾊谱梯度洗脱程序时间/min流速/(mL四min-1)A/%B/%曲线类型/0.45.095.0/10.45.095.0630.495.05.0640.45.095.01⼀⼀1为即时变化,6为线性变化1.4.2⼀质谱条件⼀电喷雾离⼦源;正离⼦扫描;多反应监测MRM;离⼦源温度150?;雾化温度500?;雾化器流速1000L/h;锥孔⽓流速150L/h;⽑细管电压2.9kV;定性⼆定量离⼦对及对应的锥孔电压和碰撞能量见表2三表2⼀7种药物定性、定量离⼦及对应的锥孔电压和碰撞能量药物定性离⼦对(m/z)定量离⼦对(m/z)锥孔电压/V碰撞能量/eV头孢氨苄348.13>157.97348.13>173.96348.13>157.972.06.012.0头孢拉定350.08>176.04350.08>157.97350.08>157.974.014.08.0头孢匹林424.09>291.98424.09>151.97424.09>291.981812.020.0克林霉素425.21>126.05425.21>377.08425.21>126.054.026.018.0头孢噻肟456.09>395.93456.09>323.97456.09>395.9322.08.012.0阿奇霉素749.41>591.39749.41>158.08749.41>591.3934.036.024.0罗红霉素837.43>679.33837.43>679.33837.43>679.3316.018.040.01.4.3⼀样品前处理⼀称取2?0.02g匀质样品,置于50mL离⼼管内,加⼄腈溶液(15+2,V/V)10mL,涡旋混匀,中速振荡10min,8000r/min离⼼5min,转移上清液于另⼀50mL离⼼管内三重复提取⼀次,合并提取液三向提取液中加正⼰烷8mL,涡旋混合5min,5000r/min离⼼5min,吸取下层溶液10mL于鸡⼼瓶中,50?下旋转蒸发⾄⼲,加⼊2.00mL20%⼄腈溶液溶解残渣,过0.22µm微孔滤膜,供液相⾊谱-串联质谱检测三2⼀结果2.1⼀线性关系⼀精密吸取200µg/L的标准混合中间液0.02⼆0.05⼆0.10⼆0.20⼆0.50⼆1.00mL,分别加⼊鸡⼼瓶中,并向其中加⼊10mL空⽩基质,从50?下快速旋转蒸发⾄⼲开始,按照样品同步处理三从⽽配制成2⼆5⼆10⼆20⼆50⼆100µg/L的系列标准溶液,并依次上机,以各药物的质量浓度为横坐标,定量离⼦的质量⾊谱峰⾯积为纵坐标,绘制标准曲线(表3),可以看出7种药物在2100µg/L范围内呈现出良好的线性关系,相关系数均在0.998以上三表3⼀7种药物的标准曲线及相关系数药物线性范围/(µg四L-1)线性⽅程相关系数r阿奇霉素2-100y=19142x+125840.9995克林霉素2-100y=108242x+894110.9999罗红霉素2-100y=48651x+140200.9991头孢氨苄2-100y=2888.9x+2879.90.9985头孢匹林2-100y=4509.4x+6.61080.9994头孢噻肟2-100y=25512x+126670.9999头孢拉定2-100y=4189.9x+1414.40.99942.2⼀检测限和定量限⼀向空⽩猪⾁⼆鸡⾁⼆鸡蛋试样中添加适量的混合标准溶液,经处理上机三根据特征质量⾊谱峰的信噪⽐S/N>3为检测限,S/N>10为定量限,检测到阿奇霉素⼆克林霉素⼆罗红霉素的检测限为0.5µg/kg,定量限为1µg/kg,头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定的检测限为2µg/kg,定量限为4µg/kg三2.3⼀⽅法的灵敏度和准确度⼀分别在猪⾁⼆鸡⾁⼆鸡蛋组织中添加4⼆8⼆20µg/kg的7种药物进⾏回收率试验,每个浓度做5个平⾏,连续测定3批,其平均回收率⼆变异系数如表4三阿奇霉素⼆克林霉素⼆罗红霉素的回收率在80%100%之间,头孢氨苄⼆头孢匹林⼆头孢噻肟⼆头孢拉定的回收率在70%90%之间,批内批间的变异系数均⼩于20%三图1为空⽩鸡⾁中添加8µg/kg的各药物特征离⼦质量⾊谱图三表4⼀7种药物的标准曲线及相关系数药物添加量/(µg四kg-1)平均回收率/%批内差异/%批间差异/%阿奇霉素4⼆8⼆2088⼆91⼆9914.8⼆7.2⼆5.816.5⼆9.9⼆9.2克林霉素4⼆8⼆2088⼆89⼆9112.4⼆4.1⼆4.516.8⼆11.3⼆8.3罗红霉素4⼆8⼆2089⼆93⼆9717.0⼆10.0⼆8.616.6⼆11.5⼆10.1头孢氨苄4⼆8⼆2075⼆79⼆8812.1⼆6.1⼆5.314.5⼆11.2⼆9.2头孢匹林4⼆8⼆2072⼆78⼆8212.2⼆5.4⼆4.111.6⼆9.5⼆8.9头孢噻肟4⼆8⼆2079⼆85⼆8912.5⼆4.2⼆3.614.9⼆10.2⼆10.6头孢拉定4⼆8⼆2077⼆79⼆877.2⼆8.0⼆6.511.1⼆9.2⼆10.2图1⼀空⽩鸡⾁中添加8µg/kg的7种药物特征离⼦质量⾊谱图3 讨论与⼩结3.1⼀⾊谱条件的优化⼀由于头孢类药物结构中含有较强酸性的羧基,在以硅胶为固定相中易出现拖尾和峰形异常的现象,因此选⽤⾼纯硅胶为基体并经端基封闭的反相⾊谱柱作为⾊谱分析柱[2]三已报道的流动相有甲酸铵⽔溶液和⼄腈[4],考虑到流动相中有机溶剂含量⼆离⼦强度以及pH值⼤⼩,都会对阿奇霉素⼆克林霉素⼆罗红霉素的离⼦化强度⼆溶解性以及在⾊谱柱上的分离产⽣影响[5]三所以,实验选⽤⼄腈和0.1%甲酸溶液为流动相,并进⾏梯度洗脱,得到了分离度以及对称因⼦均较好的峰形三3.2⼀质谱条件的优化⼀⾸先采⽤以100ng/mL的各种药物的标准溶液在ESI+的模式下进⾏母离⼦扫描,确定准确的准分⼦离⼦,然后测出相应2个⼦离⼦,并优化锥孔电压和碰撞能量等质谱参数,使其相应的响应值达到最⼤三3.3⼀提取液的优化⼀阿奇霉素⼆克林霉素⼆罗红霉素属弱碱性药物,易溶于酸性⽔溶液和极性较⼤的有机溶剂,这类药物的提取和净化主要依据其弱碱性⼆脂溶性和酸不稳定性进⾏优化[5],已报道提取溶剂有Tris缓冲液[6]⼆偏磷酸-甲醇溶液[7]⼆甲醇和⼄腈等,⽽头孢类药物由于本⾝结构带有羧基基团,所以在碱性环境中⽔溶性较好,已报道的提取液有磷酸盐缓冲盐(pH8.5)[8]⼆0.5%的冰醋酸⽔溶液[4,9],⼄腈等,本实验综合考虑了阿奇霉素⼆克林霉素⼆罗红霉素以及头孢类药物的溶解性之间的差异和相近之处,并考察⼄腈?⽔=95?5(V/V)⼆⼄腈?⽔=90?10(V/V)⼆⼄腈?⽔=80?20(V/V)⼆⼄腈?⽔=15?2(V/V)作为提取液,发现提取液为⼄腈?⽔=90?10(V/V)或者⼄腈⽐例增加时,头孢类的提取效率⾮常低,⽽当提取液为⼄腈?⽔=80?20(V/V)或者⽔⽐例再提⾼时,阿奇霉素⼆克林霉素⼆罗红霉素的提取率就⽐较低,最终确定⼄腈?⽔=15?2(V/V)溶液作为提取液三3.4⼀净化条件的优化⼀阿奇霉素⼆克林霉素⼆罗红霉素的净化⽅法常⽤液液萃取⼆固相萃取等⽅法,已报道的固相萃取柱有C18⼆HLB⼆SCX柱等[2]三头孢类药物的净化⽅法有固相萃取,已报道的固相萃取柱有OasisHLB[8]⼆XAD-2固相萃取等[9]三可见,7种药物提取液的净化柱可以采⽤HLB柱,但经HLB柱净化后头孢类药物的回收率较低,且过柱时容易堵塞,本实验考虑到样品基质特性,采⽤液液萃取的⽅法⽤正⼰烷除去脂肪等极性较⼩的杂质,省去固相萃取的步骤,使得前处理简洁经济,同时也得到了满意的实验结果三本研究通过摸索,得出⼄腈与⽔适宜⽐例的溶液⽤于同时提取7种药物,同时优化液相⾊谱条件和质谱条件,从⽽建⽴了猪⾁⼆鸡⾁⼆鸡蛋中7种药物同时检测的UPLC-MS/MS⽅法,并且其灵敏度和准确度满⾜药物残留的分析三参考⽂献:[1]⼀李富⽟.浅析⼈药兽⽤的原因⼆危害及对策[J].养殖天地,2008,12:19.[2]⼀孙雷,张骊,王树槐,等.超⾼效液相⾊谱串联质谱法对动物源⾷品中13种林可胺类及⼤环内酯类药物残留的检测[J].分析测试学报,2009,28(9):1058-1061.[3]⼀孙雷,张俪,汪霞,等.超⾼效液相⾊谱⼀串联质谱法对动物源性⾷品中13种β⼀内酰胺类药物残留的检测[J].分析测试学报,2009,28(5):576-580.[4]⼀范莹莹,其鲁,杨树民.⾼效液相⾊谱与质谱联⽤检测猪⾁中头孢类抗⽣素的残留[J].现代科学仪器,2007,6:81-88.[5]⼀李俊锁,邱⽉明,王超.兽药残留分析[M].上海:上海科学技术出版社,2002:413.[6]⼀CarmenI,RosaC,RanlonC.Determinationofmacrolideantibi?oticsbyliquidchromatography[J].JChromatogr:A,2001,910(2):285-290.[7]⼀CodonyR,CompanoR,GranadosM,etal.Determinationofmacrolideantibioticsliquidchromatography[J].JChromatogr:A,2002,959(1):285-290.[8]⼀李学民,曹彦忠,张进杰,等.⾼效液相⾊谱-电喷雾串联质谱法测定蜂蜜中5种头孢菌素[J].分析化学,2010,38(5):735-739.[9]⼀范莹莹,其鲁,杨树民.⾼效液相⾊谱-质谱联⽤法检测猪⾁中5种青霉素的残留[J].分析实验室,2007,26(12):76-79.(编辑:侯向辉)。
阿莫西林克拉维酸钾制剂中有关物质超高效液相色谱分析方法的建立研究
阿莫西林克拉维酸钾制剂中有关物质超高效液相色谱分析方法的建立研究【摘要】目的:针对阿莫西林克拉维酸钾制剂中有关物质分析构建超高效液相色谱分析方法,并对其应用效果进行评价。
方法:参照高效液相色谱分析方法构建超高效液相色谱分析方法,行方法学研究。
结果:阿莫西林克拉维酸钾制剂中有关物质超高效液相色谱分析法精密度、耐用性、准确度较高,在0.0005mg/mL~2.2mg/mL范围内具有较好线性关系(R>0.999)。
超高效液相色谱分析法与高效液相色谱分析法检查结果差值较小,杂质之间达到基线分离。
结论:超高效液相色谱分析法对阿莫西林克拉维酸钾制剂中有关物质测定具有较好应用效果,能够快速、准确检出杂质,为阿莫西林克拉维酸钾制剂杂质控制提供指导。
【关键词】阿莫西林克拉维酸钾制剂;超高效液相色谱分析法;线性范围;有关物质阿莫西林克拉维酸钾制剂对上呼吸道感染、皮肤软组织感染、尿路系统感染等具有确切疗效,临床治疗总有效率达到70%以上[1]。
随着阿莫西林克拉维酸钾制剂研究的不断深入,形成多种剂型,出现2:1~8:1等多种配比。
在此背景下,如何保证各类型阿莫西林克拉维酸钾制剂质量,成为相关人员关注与思考的重点问题。
以往所用阿莫西林克拉维酸钾制剂有关物质分析方法,时效性较差,且对色谱柱性能具有较高要求,为提高阿莫西林克拉维酸钾制剂有关物质分析效果,本文建立了超高效液相色谱分析方法,现将研究结果报道如下。
1仪器与试剂1.1主要仪器特世科技(上海)有限公司ACQUITY UPLC超高效液相色谱系统:ACQUITY UPLC system型超高液相色谱仪,Waters Empower3.0工作站;安捷伦1260 Infinity柱温箱;特世科技(上海)有限公司E2695高效液相色谱系统:WatersE2695高效液相色谱仪,Waters Empower3.0工作站;美国Milli-Q AdvantageA10超纯水系统等。
1.2主要试剂阿莫西林克拉维酸系统适用性对照品(中国食品药品检定研究院,批号为130588-201603);色谱乙腈(天津市康科德科技有限公司);磷酸二氢钾(山东鼎欣生物科技有限公司,分析纯);氢氧化钠(国药集团化学试剂有限公司,分析纯);阿莫西林系统适用性对照品(中国食品药品检定研究院,批号为130608-202005);10批阿莫西林克拉维酸钾制剂见表1。
复方阿莫西林纳米乳的高效液相色谱分析及有效期研究
复方阿莫西林纳米乳的高效液相色谱分析及有效期研究 摘要:利用高效液相色谱仪建立测定复方阿莫西林纳米乳(AMX-LH-NE)中阿莫西林(AMX)和盐酸左氧氟沙星(LH)两种主药含量的高效液相色谱(HPLC)分析方法,并确定该药物的有效期。结果表明,AMX和LH分别在0.5~50 μg/mL和5~60 μg/mL浓度范围内线性关系良好;平均回收率为(99.27±1.26)%和 (99.65±1.51)%,相对标准偏差(RSD)为1.27%和1.52%;平均保留时间为(10.22±0.13)min和(7.15±0.13)min;日内精密度RSD为1.56%和1.75%,日间精密度RSD为2.46%和2.62%。AMX-LH-NE的有效期为20个月。建立的HPLC分析方法专属性好,回收率、重复性和精密度高,可用于AMX-LH-NE制剂的主药含量测定及其质量控制。
关键词:阿莫西林;纳米乳;盐酸左氧氟沙星;高效液相色谱法;有效期 Study on Establishment of High Performance Liquid Chromatography Analytical Method for Determination of Compound Amoxicillin and Levofloxacin Hydrochloride Nanoemulsion and Its Expiration Date
Abstract: High performance liquid chromatography (HPLC) method was established for the determination of Amoxicillin(AMX) and Levofloxacin hydrochloride (LH) in the compound Amoxicillin and Levofloxacin hydrochloride nanoemulsion(AMX-LH-NE), and its expiration date by the high performance liquid chromatograph was detected. The results showed that two good linearity were respectively obtained by AMX in the range of 0.5~50 μg/mL and by LH in the range of 5~60 μg/mL, and the average recovery, relative standard deviation (RSD), average retention time, RSD of the with-in-day precision, RSD of the day-to-day precision of AMX and LH were (99.27±1.26)% and (99.65±1.51)%, 1.27% and 1.52%, (10.22±0.13) min and (7.15±0.13) min, 1.56% and 1.75%, 2.46% and 2.62%, respectively. The expiration date was 20 months. The two analytical methods of HPLC possessed good specificity, high recovery rate, repetitiveness and precision, it could be used to determine and control the principal agents in the preparation of AMX-LH-NE.