ME-S微蚀稳定剂
MEQ酸值

MEQ酸值
它是树脂中和度的参数,同一种产品直接反映树脂水溶性好坏的指标,它是槽液中含酸总量。
如果做出图来,还可以间接分析杂质离子污染状态。
电泳涂料的MEQ值=中和剂/胺值(酸值),也可用中和100g涂料固体份所需中和剂的毫克量来表示。
MEQ值的测定方法如下(仅适用于槽液):
①取10g电泳涂料槽液(精确到1mg)放入250ml烧杯中,加入50ml四氢呋喃,用电磁搅拌充分搅拌均匀。
②用0.1N氢氧化钠,3ml以/分的速度(自动或手动滴定均可)进行滴定。
③将所有测定的数据记作消耗碱的函数。
④经所测定的各点圆滑连接,用平行尺根据曲线的拐点找出曲线与拐点的两条平行切线的垂线相交二分之一点,此点即为中和点。
此点对应值即为消耗的碱量。
⑤计算:
MEQ=(V-V’)×N×100/WS
式中:
V—等当点时耗碱量(ml)
V’—四氢呋喃耗碱量(ml)
N—氢氧化钠溶液的浓度
S—试样的固体份(%)
W—试样重(g)
MEQ “离子交换容量”,即每克干树脂或每毫升湿树脂所能交换的离子的毫克当量数,meq/g(干)或meq/mL(湿);当离子为一价时,毫克当量数即是毫克分子数(对二价或多价离子,前者为后者乘离子价数)。
EGCG和EGCG—3Me对根管牙本质粘接界面稳定性的作用

EGCG和EGCG—3Me对根管牙本质粘接界面稳定性的作用将质量浓度为400 μg/mL的EGCG及EGCG-3Me添加到全酸蚀粘接剂Single Bond 2(SB2)中,制备改性粘接剂E-SB2及E3-SB2,SB2为对照组。
激光共聚焦显微镜和分光光度法检测改性粘接剂抗粪肠球菌的性能;微拉曼光谱仪检测粘接剂双键转化率;制备纤维桩粘接试件,用于即刻和老化后的微推出实验。
结果表明,改性粘接剂可以抑制粪肠球菌生物膜形成,且EGCG-3Me作用更显著;改性粘接剂与SB2的双键转化率和即刻微推出粘接强度差异无显著性(P>0.05);老化后改性粘接剂的微推出粘接强度显著高于SB2(P<0.05)。
标签:EGCG;EGCG-3Me;粘接剂;粘接稳定性纤维桩在残根、残冠的保存治疗中应用越来越广泛,已逐渐取代传统的金属桩及桩核成为主流的修复方式。
但是,近年来纤维桩修复的远期成功率成为备受广大学者和临床医生关注的问题[1,2]。
细菌感染、继发龋,以及纤维桩失去粘接力、从根管中脱落都是导致纤维桩修复失败的常见问题[3,4]。
因此,开发具有抗菌和提高根管牙本质粘接持久性的功能性粘接剂,是延长纤维桩修复体使用寿命的一项有效措施。
本研究拟通过表没食子儿茶素没食子酸酯(EGCG)及其甲基化修饰物(EGCG-3Me)对牙本质粘接剂进行功能改性,测定改性后粘接剂对根管常见致病菌、粪肠球菌的抗菌性能、粘接剂双键转化率(degree of conversion,DC)以及冷热循环老化前后粘接剂与根管牙本质的粘接性能,初步探索EGCG及EGCG-3Me功能改性的粘接剂对根管牙本质粘接界面稳定性的作用,以期为提高纤维桩远期修复效果提供可靠的科学依据。
1 实验部分1.1 主要原材料全酸蚀粘接剂Single Bond 2(N689951,3M ESPE,美国);复合树脂FiltekTM Z250(N655907,3M ESPE,美国);35%磷酸凝胶Ultra-Etch(ET4N6437,Ultradent Products,美国);脑心浸液培养基(Gibco,美国);EGCG标准品[(-)-Epigallocatechin gallate analytical standard(Sigma,美国)];EGCG-3Me(实验室制备)。
CNS号18·稳定剂和凝固剂·精简版

稳定剂&凝固剂
CNS号18.001~18.013
硫酸钙(又名石膏)
CNS 号18. 001
功能稳定剂和凝固剂、增稠剂、酸度调节剂
氯化钙
CNS 号18. 002
功能稳定剂和凝固剂、增稠剂
氯化镁
CNS 号18. 003
功能稳定剂和凝固剂
丙二醇
CNS 号18. 004
功能稳定剂和凝固剂、抗结剂、消泡剂、乳化剂、水分保持剂、增稠剂乙二胺四乙酸二钠
CNS 号18. 005
功能稳定剂、凝固剂、抗氧化剂、防腐剂
柠檬酸亚锡二钠
CNS 号18. 006
功能稳定剂和凝固剂
葡萄糖酸-δ- 内酯
CNS 号18. 007
功能稳定剂和凝固剂
刺梧桐胶
CNS 号18. 010
功能稳定剂
α- 环状糊精
CNS 号18. 011
功能稳定剂、增稠剂
γ- 环状糊精
CNS 号18. 012
功能稳定剂、增稠剂
谷氨酰胺转氨酶
CNS 号18. 013
功能稳定剂和凝固剂。
巯基苯并噻唑

巯基苯并噻唑巯基苯并噻唑英文名:2-MercaptobenzothiazoleCAS:149-30-4EINECS:205-736-8分子式: C7H5NS2分子量:167.25中文名称: 促进剂M2(3H)苯并噻唑硫酮2-巯基苯并噻唑2-硫醇基苯并噻唑2-巯基-1,3-硫氮茚促进剂MBT对甲基苯并噻苯并噻唑硫M快熟粉淡黄色单斜针状或叶片状结晶。
熔点180.2-181.7℃,相对密度1.42。
25℃时的溶解度(g/100ml):乙醇2.0,乙醚1.0,丙酮10.0,苯1.0,四氯化碳<0.2。
尚溶于冰醋酸,溶于碱和碳酸碱溶液,不溶于水。
有苦味,有不宜人的气味。
用途:2-巯基苯并噻唑作为通用型硫化促进剂,广泛用于各种橡胶。
对于天然橡胶和通常以硫磺硫化的合成胶具有快速促进作用。
但使用进需要氧化锌、脂肪酸等活化。
2-巯基苯并噻唑常与其他促进剂体系并用,如与二硫代秋兰姆和二硫代氨基甲酸碲并用可作丁基胶的促进剂、与三盐基顺丁稀二酸铅并用,可用于浅色耐水的氯磺化聚乙烯胶料。
在胶乳中常与二硫代氨基甲酸盐并用,而与二乙基二硫代氨基甲酸二乙胺并用时,可室温硫化。
该品在橡胶中易分散、不污染。
但由于其有苦味,故不宜用于食品接触的橡胶制品。
促进剂M是促进剂MZ,DM,NS,DIBS,CA,DZ,NOBS,MDB等的中间体,2-巯基苯并噻唑与1-氨基-4-硝基蒽醌和碳酸钾在二甲基甲酰胺中回流3h,可制得染料分散艳红S-GL(C.I.Disperse Red 121)。
这种染料用于涤纶及其混纺织物的染色。
2-巯基苯并噻唑用作电镀添加剂时又称酸性镀铜光亮剂M,在以硫酸铜为主盐的光亮镀铜时作为铺助光亮剂。
此外,该品还用于制取农药杀真菌剂、氮肥增效剂、切消油和润滑添加剂、照相化学中的有机防灰化剂、金属腐蚀抑制剂等。
MCM-41

Synthesis and characterization of Mn–MCM-41andZr–Mn-MCM-41M.Selvaraja,*,P.K.Sinha b ,K.L ee a ,I.Ahn a ,A.Pandurangan c ,T.G.Leea,*aYonsei Center for Clean Technology,Yonsei University,Seoul 120-749,Republic of KoreabCWMF,BARC Facilities,Kalpakkam 603102,IndiacDepartment of Chemistry,Anna University,Chennai 600025,IndiaReceived 14July 2004;received in revised form 30September 2004;accepted 5October 2004Available online 10December 2004AbstractMesoporous silica molecular sieves MCM-41containingmanganese ions (Zr–Mn-MCM-41)with Si/(Zr+Mn)ratio equal to 49,98,147,196,262and 327manganese ions with Si/Mnratio equal to 15,31,46,61,82and 102respec-tively,were synthesized under hydrothermal using cetyltrimethylammonium (CTMA +)surfactants as template in the absence of auxiliary organics.The viz.Zr–Mn-MCM-41and Mn-MCM-41were characterized using several techniques,e.g.ICP-AES,XRD,FTIR,Nitrogen adsorption,TG/DTA,ESR,UV–Vis/DRS,SEM and TEM.ICP-AES studies indicated the content of zirconium and manganese in the mesoporous materials.XRD studies indicated that the materials had the standard MCM-41structure.FTIR studies showed that zirconium and manganese ions were incorporated into the hexagonal mesoporous structure of Zr–Mn-MCM-41and Mn-MCM-41.The thermal stability of the as-synthesized materials was studied using TG/DTA.Nitrogen adsorption was used to determine specific surface area,pore diameter,pore volume and wall thickness in the calcined Zr–Mn-MCM-41and Mn-MCM-41mesoporous molecular sieves.The incorporated of zirconium and ions oxidation state was determined by ESR and UV–Vis/DRS.The morphology of Zr–Mn-MCM-41and Mn-MCM-41was determined by SEM.The inference that the Zr–Mn-MCM-41and Mn-MCM-41mesoporous materials had uniform pore was obtained by TEM studies.The incorporated metal ions as Zr 4+,Mn 2+and Mn 3+in Zr–Mn-MCM-41are coordinated to by tetrahedral,disordered octahedral and tetrahedral environments respectively.The zirconium and manganese ions are homogen-ously dispersed on the inside silica surface of the Zr–Mn-MCM-41,but only the manganese ion is not homogenously dispersed on the inside surface of the Mn-MCM-41under hydrothermal conditions.Thus the Zr–Mn-MCM-41mesoporous molecular sieves are more useful for oxidation of olefin reaction with TBHP,e.g.,in the of trans -stilbene,trans -stilbene-oxide (84.58%)was found to be the highest in the presence of Zr–Mn-MCM-41(49).Ó2004Elsevier Inc.All rights reserved.Keywords:Zr–Mn-MCM-41;Mn-MCM-41;Hydrothermal stability;Thermal stability;Lewis acidity1.IntroductionSince the discovery of MCM-41in 1992by Mobil Sci-entists [1,2],numerous studies concerning the prepara-tion conditions,synthesis mechanism,characterization and use of these materials as catalysts and catalyst sup-ports in various [3–6]have been reported.In-deed,pure siliceous MCM-41can be used in inclusion for the encapsulation of conducting quantum wires,the silicate framework providing the isolating part of the device.But as these are pure silica materials,lacking sufficient acidity,it is difficult to use them directly as catalysts.Metallosilicate1387-1811/$-see front matter Ó2004Elsevier Inc.All rights reserved.doi:10.1016/j.micromeso.2004.10.004*Corresponding authors.Tel.:+82221235751/409941;fax:+8223126401.E-mail addresses:selvarajman25@ (M.Selvaraj),ted-dy.lee@yonsei.ac.kr (T.G.Lee)./locate/micromesoMicroporous and Mesoporous Materials 78(2005)139–149molecular sieves,obtained by isomorphous substitution of Si by metal ions in silicate structures,exhibit catalytic activity in selective oxidation reactions using hydrogen peroxide and alkylhydroperoxides at mild conditions.In recent years,attention has increasingly been directed towards the study of metal-containing mesoporousM41S type molecular sieves with large pores (20–100A˚diameter)suitable for the transformation of bulky or-ganic compounds [1,2,7–13].Metal ions such as Al [14–17],Ti [6,18],Fe[20–25],V [26,27],and Ga[28–30]have been incorporatedinto the MCM-41et al.[31–33]reported the details of synthesis and characterization of bimetal-lic as Zn-Al-MCM-41mesoporous materials.How-ever,hydrothermal stability,hydrophobicity,thermal stability and acid strength in the mesoporous materials increased by the introduction of the incorporation of zinc.Manganese are well known catalysts for epoxidation reactions [34].Recently,many research groups worked on the immobilization of Mn complexes onto the mesoporous material.Although ion-exchange method was applied to Al-MCM-41[35],only lower loading amount can be obtained by this method.Caps and Tsang [36]prepared Mn-MCM-41with a molecular organic chemical vapor deposition (MOCVD)method.Burch et al.[37]prepared surface-grafted manganese–oxo species on the walls of MCM-41channels.Yonemi-tsu et al.[38]firstly developed the template ion-exchange (TIE)method for the synthesis of Mn-MCM-41,and they argued that manganese was highly dispersed in the mesopore of MCM-41and only existed as Mn 2+.But,when the other transition metal ions are incorporated in Mn-MCM-41,the environment struc-ture,morphology and distribution of the Mn in the framework and also hydrothermal stability have not established.In the present study,Zr–Mn-MCM-41and Mn-MCM-41materials with different silicon to metal/metals ratio have been synthesized using hydrother-mal method.The materials characterized by ICP-AES,XRD,FTIR,Nitrogen adsorption,TG/DTA,DR/UV–Vis spectroscopy,ESR,SEM and TEM.Mesostructure,metal incorporation,morphology of Zr–Mn-MCM-41,the site geometry,coordination and distribution of Mn in Zr–Mn-MCM-41when the Zr-ions incorporated in Mn-MCM-41are subsequently investigated.2.Experimental 2.1.MaterialsThe syntheses of Zr–Mn-MCM-41and Mn-MCM-41materials were carried out by hydrothermal method using sodium metasilicate (Na 2SiO 3Á5H 2O),Zirconiumtetraisopropoxide ((Zr–(O–CH–(CH 3)2)4),Manganese (II)acetate ((CH3COO)2Mn),cetyltrimethylammo-niumbromide (C 3)3N +Br)and sulfuric acid (H 2SO 4).The all were purchased from M/s Aldrich &Co.,2.2.Synthesis of Zr–Mn-MCM-41and Mn-MCM-412.2.1.Synthesis of Zr–Mn-MCM-41The Zr–Mn-MCM-41was synthesized according to the previous report [39]using hydrothermal method.For the synthesis of the Zr–Mn-MCM-41(Si/Zr +Mn =49),21.2g (1mol)of sodium metasilicate (44–47%SiO 2)dissolved in 50g of deionised water was with 0.54g (0.015mol)of zirconium (IV)isoprop-and 0.4g (0.015mol)of manganese (II)acetate (dissolved in 10g of deionized water)solution.This mix-ture was stirred for 30min using a mechanical stirrer at a speed of about 250rpm and in order to reduce the pH to 10.8,1N of sulphuric acid was added with continuous stirring for another 30min at a speed of about 250rpm until the gel formation.After that,9.1g (0.25mol)of cetyltrimethylammonium bromide was added drop by drop (30ml h À1)through the dual syringe pump so that the gel was changed into suspension.After further stirring for 1h the resulting synthesis gel of composition 1SiO 2:0.015ZrO 2:0.015MnO:0.25CTMABr:100H 2O,was transferred into Teflon-lined steel autoclave and heated to 165°C for 48h.After cooling to room temper-ature,the material was recovered by filtration,washed with deionised water and ethanol and finally calcined in flowing air at 540°C for 6h.The different Zr–Mn-MCM-41catalysts (Si/Zr +Mn =98,147,196,262and 327)were also synthe-sized in a above similar manner wherein only the ratio of sodium metasilicate,zirconium (IV)isopropoxide and manganese (II)acetate was adjusted and the input in gel molar compositions 1SiO 2:x ZrO2:y MnO:0.25CT-MABr:100H 2O;(x =0.008–0.0025,y =0.008–0.0025).2.2.2.Synthesis of Mn-MCM-41The Mn-MCM-41catalysts (Si/Mn =15,31,46,61,82and 102)were also synthesized in a above similar manner wherein only the ratio of sodium metasilicate and manganese (II)acetate was adjusted without zirco-nium source and the input in gel molar compositions 1SiO 2:x MnO:0.25CTMABr:100H 2O;(x =0.033–0.005).2.3.Characterization2.3.1.ICP-AESThe zirconium and manganese content in Zr–Mn-MCM-41and Mn-MCM-41were recorded using ICP-AES with allied analytical ICAP 9000.140M.Selvaraj et al./Microporous and Mesoporous Materials 78(2005)139–1492.3.2.XRDThe crystalline phase identification and phase purity determination of the calcined Zr–Mn-MCM-41and Mn-MCM-41samples were carried out by XRD(Phi-lips,Holland)using nickelfiltered Cu K a radiation (k=1.5406A˚).The samples were scanned from0.5°to 5°(2h)angle in steps of0.5°,with a count of5s at each point.In order to protect the detector from the high en-ergy of the incident and diffracted beam,slits were used in this work.2.3.3.FTIRInfrared spectra were recorded with a Nicolet Impact 410FTIR Spectrometer in KBr pellet(0.005g sample with0.1g KBr)scan number36,resolution2cmÀ1. The data was treated with Omnic Software.2.3.4.N2-AdsorptionThe surface areas and pore properties of Zr–Mn-MCM-41and Mn-MCM-41samples before and after hydrothermal treatments were analyzed using a NOVA-1000(QUANTACHROME,version5.01)sorp-tometer.The calcined samples were dried at130°C and evacuated overnight for8h inflowing arogn atflow rate of60ml minÀ1at200°C under vacuum.Surface area, pore size,pore volume and wall thickness was obtained from these isotherms using the conventional BET and BJH equations.2.3.5.TG-DTAThe weight loss,dehydration and dehyoxylation for as-synthesized Zr–Mn-MCM-41and Mn-MCM-41 samples was evaluated by a Thermogravimetric-Differ-ential Thermal Analysis(TG-DTA)in a Rheometric scientific(STA15H+)thermobalance.10–15mg of as-synthesized MCM-41was placed in a platinum pan and heated from room temperature to1273K at a heat-ing rate of2K/min in air withflow rate of50ml/min. For comparison experiments,samples were dried at 323K for the same period.The data were collected at 30-s intervals using on-line PC.2.3.6.ESRFor ESR measurements,Zr–Mn-MCM-41and Mn-MCM-41samples were loaded into2mm i.d x150mm length suprasil quartz tubes.ESR spectra were recorded at X-band at either293or77K on a Bruker ESP300 spectrometer.The magneticfield was calibrated with a Varian E-500gaussmeter.The microwave frequency was measured by a Hewlett Packard HP5342A fre-quency counter.2.3.7.UV–visible/Diffuse reflectance spectroscopyThe diffuse reflectance UV–visible spectra were meas-ured with a Perkin-Elmer330Spectrometer equipped with a60mm Hitachi integrating sphere accessory.Powder samples were loaded in a quartz cell with Supra-sil windows,and spectra were collected in the200–800nm wavelength range against quartz standard.2.3.8.SEMThe SEM microscope of a typical sample of MCM-41 materials were obtained on a JEOL2010microscope operated at200KV from a thinfilm dispersed on a holly carbon grid in ethanol solvent.2.3.9.TEMTEM images were recorded using a JEOLJEM-200CX electron microscope operating at200KV with modified spectrum stage with objective lens parameters C s=0.41mm and C c=0.95mm,giving an interpretable point resolution of Ca.0.185nm.Samples for analysis were prepared by crushing the particles between two glass slides and spreading them on a holly carbonfilm supported on a Cu grid.The samples were briefly heated under tungstenfilament light bulb in air before transfer into the specimen chamber.The images were recorded at magnifications of100000·.3.Results and discussion3.1.ICP-AESThe zirconium and manganese ions content in Mn-MCM-41and Zr–Mn-MCM-41have been observed using ICP-AES.The observed zirconium and manga-nese ions content in the samples have been presented in Table1.3.2.XRDFig.1a and b show the X-ray powder diffraction pat-terns of calcined Mn-MCM-41and Zr–Mn-MCM-41 samples having different ratio of Si/Mn and Si/Zr+Mn respectively.The X-ray diffractograms of Zr–Mn-MCM-41and Mn-MCM-41after calcinations in air at540°C for6h, contain,a sharp d100reflection line in the range 2h=2.15–2.30°and 2.03–2.22°respectively.Physico-chemical properties of these mesoporous materials are summarized in Table1.For example,the pore-to-pore distance of Zr–Mn-MCM-41could be determined by the XRD patterns.The XRD patterns of calcined Zr–Mn-MCM-41(49)with characteristic peaks of hexago-nal(p6mm)symmetry and with d100of41.090are shown in Fig.1b.The repeating distance(a0)between pore cen-ters was47.440.The hexagonal unit cell parameter(a0) was calculated using the formula as a0=2d100/p3from d100,which was obtained from the peak in the XRD pat-tern by BraggÕs equation(2d sin h¼k,where k=1.54060for the Cu K a line).The value of a0wasM.Selvaraj et al./Microporous and Mesoporous Materials78(2005)139–149141equal to the internal pore diameter plus one pore wall thickness.These peaks reflections were indexed based on the hexagonal unit cell as described by Beck et al.[2].The XRD peaks shifted to lower 2h values for the rest of the samples.The higher d -spacing and unit cell parameter values observed with increasing metal content suggest an incorporation of metal in the framework locations [40]because of its (Zr–O ($1.71160)and Mn–O ($1.73080))longer bonding length with oxygen than Si–O ($1.5090).However,there is no regular rule in MCM-41as it has an amorphous structure where both bond length and angle may be change.The pertur-bation of d-spacing and unit cell parameter is hypothe-sized to be dependent on the metal substitution stateresulting from the sample preparation method,i.e.,coordination number of metal with OH groups and also along with bond length and bond angle of metal ion on the silica surface.3.3.FTIRInfrared spectroscopy had been used extensively for the characterization of transition-metal cation modified zeolites.The as-synthesized Zr–Mn-MCM-41and Mn-MCM-41samples exhibit absorption bands around 2921and 2851cm À1corresponding to n -C–H and d -C–H vibrations of the surfactant molecules.The broad bands around 3500cm À1may be attributed surfaceTable 1Physicochemical characterization of Zr–Mn-MCM-41and Mn-MCM-41Catalysts Zr content d (wt%)Mn content d (wt%)d -spacing a (A˚)Unit cell parameter aa 0(A ˚)Surface areab (m 2/g)Pore size bD (A ˚)Pore volume b (cm 3/g)Wallthickness c (A ˚)Zr–Mn-MCM-41(49)0.1650.16641.0947.4483530.30.8117.14Zr–Mn-MCM-41(98)0.0830.08240.5646.8386330.20.7916.63Zr–Mn-MCM-41(147)0.0520.05540.1546.3689030.10.7516.26Zr–Mn-MCM-41(197)0.0410.04139.4345.5292429.90.7015.62Zr–Mn-MCM-41(261)0.0320.03138.4044.3494329.80.6914.54Zr–Mn-MCM-41(327)0.0230.02536.8142.50102529.40.6813.1Mn-MCM-41(15)–0.33043.5150.2486332.70.9917.54Mn-MCM-41(31)–0.16542.6149.2088432.60.9816.60Mn-MCM-41(46)–0.11142.2648.7990332.50.9716.29Mn-MCM-41(61)–0.08241.6148.1194532.40.9415.71Mn-MCM-41(82)–0.06240.7147.0099932.30.9114.7Mn-MCM-41(102)–0.05039.7945.94103432.20.8913.74a Values obtained from XRD studies.b Values obtained from N 2-adsorption results.c Wall thickness (t )=Unit cell parameter(a 0)pore size(D ).dThe results obtained from ICP-AES.142M.Selvaraj et al./Microporous and Mesoporous Materials 78(2005)139–149silanols and adsorbed water molecules,while deforma-tional vibrations of adsorbed molecules cause the absorption bands at1623–1640cmÀ1[41](the results of peaks have not shown infigures).The substitution of silicon by zirconium and manganese causes shifts of the lattice vibration bands to lower -pared to the Si-MCM-41,the wave number of the anti-symmetric Si–O–Si vibration band of Mn-MCM-41and Zr–Mn-MCM-41samples decreases to1086and 1080cmÀ1,receptively(Fig.2a and b).Theses shifts should be due to the increase of the mean Si–O distance in the walls caused by the substitution of the small sili-con(radius40pm)by the larger size of Mn2+(radius 83pm)and Zr4+(radius59pm)[42].The observed shifts, which depend as well on the change in the ionic radii as on the degree of substitution,are comparatively small. Therefore,only a low degree of substitution is sug-gested.Interestingly,the wavenumber shifts decrease in the series Zr–Mn-MCM-41>Mn-MCM-41, although the ionic radius of zirconium is smaller than those of manganese.While the wave number of the anti-symmetric Si–O–Zr and Si–O–Mn vibration bands of Zr–Mn-MCM-41and Mn-MCM-41samples de-creases to957and962cmÀ1,receptively(Fig.2a and b).The absorption band at1057and1223cmÀ1are due to asymmetric stretching vibrations of Si–O–Si bridges,while the960to965cmÀ1bands are due to Si–OÀM+(M=Mn and Zr)vibrations in metal incorporated silanols.By the disappearing peaks at 2851and2921cmÀ1,one could conclude that calcination of the original framework was complete and the identity of organic molecule completely disappeared from the calcined M-MCM-41.Upon introduction of higher metal content,most of the bands shifted to higher wave numbers,consistent with their incorporation in lattice positions.Addition-ally,an absorption band in the range962–967cmÀ1as-signed to a stretching vibration of Si–OÀM+linkage was observed.This is generally considered to be a proof of the incorporation of the heteroatom into the frame-work.Cambler et al.[43]have reported similar stretch-ing vibrations of Si–OH groups present at defect sites.3.4.N2-adsorption isothermFig.3a and b show the isotherm of nitrogen adsorp-tion by calcined Mn-MCM-41and Zr–Mn-MCM-41 measured at liquid nitrogen temperature(77K).Three well-defined stages may be identified:(1)A slow increase in nitrogen uptake at low relative pressure,correspond-ing to monolayer-multilayer adsorption on the pore walls;(2)a sharp step at intermediate relative pressures indicative of capillary condensation within mesopores; and(3)a plateau with a slight inclination at high relative pressures associated with multilayer adsorption on the external surface of the crystals[44].A fourth stage,M.Selvaraj et al./Microporous and Mesoporous Materials78(2005)139–149143characterized by a sharp rise in N2uptakefilling all other available pores as the pressure reaches saturation (p/p0=1.0),may be identified in some isotherms.Each isotherm showed type IV character,which is a typical shape for mesoporous MCM-41.The third stage of the isotherm shifts slightly toward higher relative pressure with the incorporation of transition metal(Zr+Mn and Mn).The hysteresis loop broadened with an in-crease in the metal content suggesting some disorder in the pore system arising from metal incorporation.Incor-poration of metal in MCM-41has significant effect on these parameters.The surface area values decreased with increasing metal content.The pore diameter,pore vol-ume and wall thickness increased in Zr–Mn-MCM-41144M.Selvaraj et al./Microporous and Mesoporous Materials78(2005)139–149and Mn-MCM-41with increasing metal content.and are listed in Table1.In the crystalline zeolites,metal incorporation slightly increases the pore size because of its longer bonding length with oxygen than Si–O. However,there is no regular rule in MCM-41as it has an amorphous structure where both bond length and angle may ually,it has been observed that the pore size of MCM-41is decreased after metal incor-poration,but there is no clear explanation for this obser-vation.MCM-41has thin pore walls relative to zeolites so that the incorporated metal cannot be substituted into the silica framework completely.That is,a part of the metal will be exposed on the pore wall surface so that it might have properties similar to impregnated metal complexes on the MCM-41walls.The incorpo-rated metal may interact with surface hydroxyl groups and may contract the pore wall when combined with two or three hydroxyl groups,so that the pore size will be de-creased[45,46].If these metals are incorporated deep within the silica framework,this pore shrinkage might not occur as with condensed surface hydroxyl groups, and the pore size might be increased instead by the in-creased metal-oxygen bond length,as for zeolites.In this study,we obtained an increased pore size after both metal as Zr and Mn,and alone Mn incorporation as shown in Fig.2a and b respectively.This is an unusual result, and strongly suggests that Zr and Mn are incorporated into silica framework according to the above hypothesis. This can be confirmed indirectly by the ease of metal reduction,and such a result has been reported[47].The remarkable improvement of hydrothermal stabil-ity due to the higher metal incorporation is further evi-denced by the measurement of N2adsorption.The surface areas of all the MCM-41materials were mea-sured after they had been treated in boiling water for1 week.The surface areas slightly decreased from863–1034to830–980m2/g in the Mn-MCM-41samples and also slightly decreased from1025-890to967–1002m2/g in Si/(Zr+Mn)ratios of147–327in Zr–Mn-MCM-41 samples,but the surface area does not decrease in other Zr–Mn-MCM-41samples.The Si–O–Zr and Si–O–Mn bonds are relatively stable to further attack from boiling water[39,48];the presence of Mn2+and Zr4+creates a negative charge on the surface of the pore walls,repel-ling OHÀions and therefore preventing the hydrolysis of siloxane bonds and also resulting in an increase in the number of acid sites.The surface area in the Mn-MCM-41and Zr–Mn-MCM-41(Si/Zr+Mn=147–327) samples decreased due to low metal incorporation/non-framework as it does not create sufficient negative charges on the surface of the pore walls.But the surface area of other Zr–Mn-MCM-41samples does not decrease due to repelling OHÀions higher on the pore walls.Hence,it is concluded that Zr is irreversibly incor-porated into the structure of Zr–Mn-MCM-41samples. Thus,the hydrothermal stability is higher in Zr–Mn-MCM-41(49)than that in other Zr–Mn-MCM-41and Mn-MCM-41samples.3.5.Thermal analysisThermogravimetric analysis of the catalysts show dis-tinct weight losses that depend on framework composi-tion.Representative thermograms are given in Fig.4a (Mn-MCM-41)and Fig.4b(Zr–Mn-MCM-41).Gene-rally,when the metal content increases,there is a de-crease in organic content and increase in water content.Three distinct regions of weight loss are noted in the temperature range50–150°C,150–350°C and 350–550°C.Thefirst weight loss($4.6–16.61%)corre-sponds to the desorption and removal of the water and/or ethanol molecules physisorbed on the external surface of the crystallites or occluded in the macropores and mesopores present between the crystallites aggre-gates.A second weight loss($38.58–40.5%),between 150°C and350°C is attributed to the removal of the or-ganic template.Finally,a third weight loss($1.51–1.9%) between350°C and550°C is related to water loss from the condensation of adjacent silanol groups to form siloxane bond[49].The total weight loss at1000°C of the Mn-MCM-41and Zr–Mn-MCM-41samples are in the44.69–59.01%range.However,the distribution of successive weight loss depends on the framework or sub-stituted silicon to metal ratio[50].Thus,weight loss was higher in the low metal contents of MCM-41materials than that in the high metal contents of MCM-41 materials.3.6.ESRThe Q-band ESR spectrum of synthesized Mn-MCM-41at150°C in the presence of suphuric acid exhibits a typical Mn2+spectrum,sextet lines with a hyperfine coupling(A)of81G and g=2.00,as shown in Fig.5a.Low intensity forbidden transitions are evi-dent in room temperature spectra,indicating that the Mn2+ions are rather incorporated.The greater incorpo-ration of the Mn2+species in as-synthesized Mn-MCM-41at150°C can be attributed either to its location within the wall or in the interface region of the polar head groups of the template and the silica walls.How-ever,their high mobility indicates that they are not located within the inorganic walls but are within the pores.This indicates that the Mn2+is more mobile after calcination.The same spectrum was obtained from cal-cined Mn-MCM-41that did not exhibit Mn2+ESR sig-nals prior to calcinations.From the above results,it is inferred that the Mn2+is coordinated to Si(IV)by dis-torted octahedral environments.The preliminary EPR analysis was carried out for the Zr–Mn-MCM-41.A strong resonance at g=2.01with a hyperfine coupling of91g was observed(Fig.5b)whichM.Selvaraj et al./Microporous and Mesoporous Materials78(2005)139–149145could be easily attributed to a distorted octahedral Mn2+species.The hyperfine lines are slightly broader for Zr–Mn-MCM-41than for Zr-MCM-41.The spin Hamilton parameters are also similar to those reported by Piaggio et al.[51]for the homogeneous Mn(II)-salen complex,suggesting the incorporation of the Zr and Mn in the MCM-41materials.3.7.Diffuse reflectance UV/visible spectroscopyThe calcined Mn-MCM-41samples are characterized by reflectance UV–Visible spectroscopy.Two bands at 270and500nm were observed for these samples.The A1g!T2g crystalfiled transitions of Mn2+in Mn2O3 or MnO showed a band at500nm[52].Thus the band at500nm was assigned to Mn2+probably existing on the surface of MCM-41.However,the band at270nm with such a high intensity was not reported in the liter-ature.The charge transfer transition of O2À!Mn2+in Mn3O4in which Mn was disordered octahedrally coor-dinated with oxygen exhibited a band at320nm[52]. We thus tentatively assigned the intense band observed at lower wavelength(270nm)to the charge transfer transition of O2À!Mn3+tetrahedral coordination,i.e in the framework of MCM-41.From the above results, it is inferred that the Mn2+and Mn3+ions may be coor-dinated with Si(IV)by disordered octahedral and tetra-hedral environments respectively.The DR UV–Vis spectra of Zr–Mn-MCM-41were found to be similar for those of Zr-MCM-41[39]and Mn-MCM-41in that the peaks were in the same position and the Zr4+and Mn2+are coordinated to Si(IV)by tet-rahedral and disordered octahedral environments respectively.From the ESR and UV–Vis/DRS results, theoretically,we conclude that the Lewis acidity in Zr–Mn-MCM-41materials are higher than that that in Mn-MCM-41due to the higher electron-pair donated to the inner side silica surface.3.8.SEMAll Mn-MCM-41and Zr–Mn-MCM-41materials are having micellar rod-like shape hexagonal or spherical edges,as shown in the Fig.6a(Mn-MCM-41(31))and Fig.6b(Zr–Mn-MCM-41(49)).Each rod,itself trans-formed into the MCM-41hexagonal-phase mesostruc-ture.Because all materials have been synthesized using cetyltrimethyammonium bromide as surfactant in the liquid crystal template mechanism,Steel et al.[53]pos-tulated that CTMABr surfactant molecules assembled directly into the hexagonal liquid crystal phase upon addition of the silicate species,based on14NMR spectro-scopy.The size of micellar rod like shape and hexagonal phase in Zr–Mn-MCM-41materials are slightly smaller than those in Mn-MCM-41.3.9.TEMThe TEM image of all Mn-MCM-41and Zr–Mn-MCM-41samples(Fig.7a(Mn-MCM-41(31))and Fig.7b(Zr–Mn-MCM-41(49))exhibit ordered hexago-nal arrays of mesopores with uniform pore size[1]. The corresponding electron diffraction pattern also shows reflections contrast in the TEM image of the sam-ple,the distance between mesopores are estimated,in good agreement with the value determined from XRD and N2-adsorption measurements values.Both pore channels and hexagonal symmetry can be clearly identified in the TEM image for the Mn-MCM-41and Zr–Mn-MCM-41samples which indicates that the MCM-41materials have only one uniform phase as inferred from the XRD results.Interestingly,the TEM image viewed down the d110direction shows that pore channels are not straight,they are arc-like.In con-trast,the TEM image recorded along the d110direction of MCM-41showshorizontal.Fig.6.(a)SEM image of Mn-MCM-41(31).(b)SEM image of Mn-MCM-41(49).M.Selvaraj et al./Microporous and Mesoporous Materials78(2005)139–149147。
SAG-005(抗水解稳定剂)
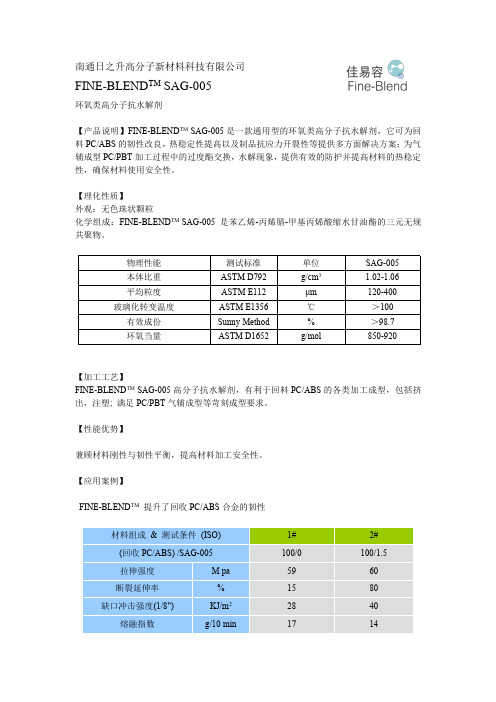
南通日之升高分子新材料科技有限公司FINE-BLEND TM SAG-005环氧类高分子抗水解剂【产品说明】FINE-BLEND TM SAG-005是一款通用型的环氧类高分子抗水解剂,它可为回料PC/ABS的韧性改良,热稳定性提高以及制品抗应力开裂性等提供多方面解决方案;为气辅成型PC/PBT加工过程中的过度酯交换,水解现象,提供有效的防护并提高材料的热稳定性,确保材料使用安全性。
【理化性质】外观:无色珠状颗粒化学组成:FINE-BLEND TM SAG-005是苯乙烯-丙烯腈-甲基丙烯酸缩水甘油酯的三元无规共聚物。
物理性能测试标准单位SAG-005本体比重ASTM D792g/cm3 1.02-1.06平均粒度ASTM E112μm120-400玻璃化转变温度ASTM E1356℃>100有效成份Sunny Method%>98.7环氧当量ASTM D1652g/mol850-920【加工工艺】FINE-BLEND TM SAG-005高分子抗水解剂,有利于回料PC/ABS的各类加工成型,包括挤出,注塑;满足PC/PBT气辅成型等苛刻成型要求。
【性能优势】兼顾材料刚性与韧性平衡,提高材料加工安全性。
【应用案例】FINE-BLEND TM提升了回收PC/ABS合金的韧性材料组成&测试条件(ISO)1#2#(回收PC/ABS)/SAG-005100/0100/1.5拉伸强度M pa5960断裂延伸率%1580缺口冲击强度(1/8")KJ/m22840熔融指数g/10min1714***数据来源:海外知名改性公司FINE-BLEND TM 提升了回收PC/ABS 合金的熔接线强度-0.50.0.51.01.52.0344wi e l d l i n e s t r e n g t h (M p a )S A G -005 C o n t e n t (%)熔接线是注塑成型制品最严重的成型缺陷之一,它不但影响制品的外观质量,而且熔接线对制品强度有影响。
受阻胺类光稳定剂
b i r(IL )a t d cd i ee p ai o esnr sce et o A Swt o e dte 。 iz rA S r i r ue ,wt t m hs nt ye i i f sf H L i t r d ivs le eno hh s h g t c h h a i Kew rs i ee mi ; i t t izr ehns A pi f n y od :H n rdA n Lg a le;M cai d e h Sb i m; p l ao ci
量已 占稳 定 剂 总 量 的 4 % ,跃 居 各 稳 定 剂 之 首 。进 4
12 分解 过氧化 物 作 用 .
入 2 世纪 9 年代以来 ,随着受阻胺 的抗热氧稳定化 o 0 功能的开发 ,使其又有了新的应用领域_4 3 ,。 J
过氧化物的存在和积累是引发聚合物光氧化降解 的 根源。受阻胺能有效的分解过氧化物,使之转化为相对 稳定的醇、酮化合物 , 从而抑制聚合物的降解_8 7J ,。
注 。常用 的降低受 阻胺 碱 性方法 是 利用取代 基 或空 间 位阻保 护 碱性 氮 ,如 Ⅳ 烷 基 化 ,N一 氧基 化 ,Ⅳ一 一 烷 酰 基化基 团。 实 验 证 实 ,Ⅳ一 H受 阻 哌 啶 化合 物 的碱 性 最 大 ,Ⅳ一 取代烷 基 次 之 , Ⅳ 取 代 烷 氧 基 最 小 。汽 巴 一
EME 性能及成型技术简介
High Purity Fillers for Memory Device to Reduce Filler induced Soft Error. 高纯度填料用于防止存储器时出现软体错误。
CCP
Crystalline silica
Fused silica
長
春
Item CTE Thermal conductivity
a.锭粒预热Preheating of Pellet
锭粒间留有适当间隙,保证预热温度均匀.
CCP
長
春
锭粒与电极板之间距离保持3-5mm,尽量不要通过调整极板高度来调 节预热温度。
Unit E-7/degC E-4cal/ cm·sec·degC
Crystalline 50 300
Fused 5 30
CTE @ Filler type&content
CCP
長
春
结晶型硅砂(crystal silica)
CCP
長
春
熔融型硅砂(fused silica)
CCP
長
春
球形硅砂(spherical silica)
11
High filler Loading Combination of sharp distribution Filler
CCP
長
春
Image
R 0.414R 0.225R Porosity
100
29.5%
ቤተ መጻሕፍቲ ባይዱ
91.5 6.5 1.85 19.0%
12
EME固化(交联)程度行为曲线
CCP
長
春
温度
EME是一种热固性材料,在一定温度条件下会发生不可逆的交联反应。因此在 生产、储存、使用环节必须注意温度及时间控制,防止材料变质。
以极软水为补充水的循环冷却系统水质稳定剂
-1)图1 A3在剂量35mg/L的腐蚀速度倍浓缩水中的腐蚀速度及浓缩水质μS•cm-1)pH值1(2Ca2+/mmol•L-1)碱度(mmol•L-1)4098.80 4.04 3.69空白 M-1(35) M-1(50) M-2(35) M-2(50) M-3(35) M-3(50) M-4(35) M-4(50) M-5(35)M-5(50) M-6(35) M-6(50) M-7(35) M-7(50)图2 A3使用M-1~M-7腐蚀挂片后形貌2.3 Cl-剂量试验结果估计投加活性二氧化氯杀生后循环冷却水量最大值为80mg/L,因此,进行了Cl-剂量试验。
在无阀滤池清水的4.15倍浓缩水中(Cl-为80mg/L)蚀速度测定结果及浓缩水质的测定结果见表4,腐蚀药剂表4 TP304空白 M-2(8) M-2(11) M-2(14)M-2(17) M-3(11) M-3(14) M-3(17)图3 A3使用M-3、M-4腐蚀挂片后形貌空白 M-2(5) M-2(8) M-2(11)M-2(14) M-36(8) M-3(11) M-3(14)4 TP304使用M-3、M-4腐蚀挂片后正面形貌挂片试验后形貌代表见图5;TP304在无阀滤池清水4.38浓缩水中(Cl -为80mg/L)的腐蚀速度及浓缩水质的测定结果见表5,腐蚀挂片试验后形貌代表如图所示。
结论(1)较佳药剂:根据筛选结果(图1和图2)和筛选原则,7种药剂中以M-3、M-4缓蚀效果最好;(2)适宜剂量:根据剂量试验结果(表2和表)空白 M-2(8) M-2(11) M-2(14) M-2(17) M-3(11) M-3(14) M-3(17)5 A3在80mg/LCl -、4.15倍浓缩清水中腐蚀挂片后形貌M-2、M-3在循环冷却水中的适宜剂量为,相当于补充水中剂量为8~11mg/L 影响:根据Cl -剂量试验结果(表,使用药剂M-2、M-3后,即使循环冷却水浓度达到80mg/L ,A3、TP304耐蚀性也能满足GB 50050-2007《中华人民共和国国家标准冷却水处理设计规范》要求。
Mesalabs-EZS6生物指示剂说明书
Mesalabs-EZS/6生物指示剂说明书型号:EZS/6菌种:嗜热芽孢脂肪杆菌7953(1)生物指示剂用途:蒸汽灭菌培养:EZTest 培养基,55-60℃培养。
所提供的细菌培养基符合促生长能力的需求。
纯度:使用标准平板计数技术时没有污染的证据生产批号:S-459 生产日期:2013年12月19日失效日期:2015年12月19日热冲击计数:2.2×106孢子量/支载体尺寸:6mm×19mm抗力分析: D值(2)存活时间杀灭时间121℃蒸汽 2.2 9.71 23.12(3)Z值:16.9℃使用指导:注意:灭菌之后,玻璃安瓿内的生物指示剂热且带有压力。
应至少冷却10分钟。
冷却少于10分钟安瓿可能破碎以及热的液体所导致的伤害。
A 灭菌过程使用生物指示剂1.从包装盒中取适量的指示剂。
2.从标签上鉴别生物指示剂的信息。
3.将生物指示剂放在代表性装载的合适测试包装中。
4.将测试包装放置在灭菌器的最大挑战区域,通常是在靠近门排水口上方的架子上。
如果没有使用测试包,将生物指示剂放置在装载的水平位置。
5.按正常过程进行装载。
6.将带有生物指示剂的包装从灭菌器移除后应给与足够的时间进行冷却,至少10分钟以上。
7.将生物指示剂从测试的装载中取出。
8.灭菌后当标签上的化学指示剂由蓝色变为黑色。
与没有进行灭菌的生物指示剂进行区别。
注意:黑色不能表明接受过灭菌。
B 培养55-60℃的条件将满足指示剂的培养条件。
为了使指示剂解触培养基,将指示剂以垂直位置放置在塑料破碎器里面。
轻轻地挤压破碎器破坏玻璃安瓿。
将与培养基接触的生物指示剂放置在培养箱的架子上,立即进行培养。
C 解释说明:1.在培养至8小时,12小时,18小时和24小时检查生物指示剂的颜色变化。
出现黄色表明有细菌生长。
没有变化表明经过了适当的灭菌。
2.一旦发生阳性试验(变为黄色)第一时间应记录下来。
通知医院人员(例如疾病控制中心)。
3.建议的培养时间是24小时(符合US FDA/RIT协议)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ME-S微蚀稳定剂
简介:ME-S微蚀稳定剂是属于H
2SO
4
/ H
2
O
2
体系的稳定剂,用于铜箔氧化前
的表面处理,能形成均匀的、微粗化表面,为氧化提供理想的微观表面,从而达到最佳的粘合力。
一、特点
1、板面光亮,效果均匀一致;
2、微蚀速率恒定,操作方便;
3、铜溶量大,高含铜状态下,微蚀速率稳定。
二、溶液的配制
微蚀缸: H
2SO
4
(98%): 10%
H
2O
2
(50%): 5%
ME-S: 8%
DI水: 余量
(加硫酸时应慢加入并搅拌,待温度低于350C ,再加其他成分)
三、操作条件
最佳值控制范围
微蚀缸: H
2SO
4
: 80ml/L 50-100ml/L
H
2O
2
(50%):45ml/L 30-60ml/L
时间: 1min 0.5-1.5min
温度: 30 0C 25-35 0C
铜离子: <50g/L
四、药水添加与换缸
每100平方米,补加硫酸3升,50%双氧水2升和1升的ME-S稳定剂;当铜离子浓度 > 45g/L时,换缸.
五、化学分析
(1)H
2SO
4
的分析
(A)试剂:
1、1.0N的NaOH溶液
2、甲基橙指示剂
( B ) 方法:
1、精确吸取2.0ml样品于250ml锥形瓶中
2、加入50ml的DI水
3、加入 3滴甲基橙指示剂
4.、用1.0N的NaOH溶液滴定至溶液由红变黄
(C)计算:
H 2SO
4
(ml/L) = 27 ×(V . N) NaOH / Vs
(2)H
2O
2
的分析
(A)试剂:
1、0.1N的KMnO
4
标准液(3.16g/L)
2、20%的H
2SO
4
(B)方法:
1、精确吸取0.5ml样品于250ml锥形瓶中
2、加入30ml D.I水
3、加入20ml 20%的H
2SO
4
4、用0.1N KMnO
4
滴定至红色30s不消失,即为终点。
记录滴定体积。
(C)计算:
H 2O
2
(50%, ml/L) = 28.4× (V . N) KMnO
4
/ Vs
(3)Cu2+的分析
(A)试剂:
1、浓NaOH液
2、PAN指示剂
3、pH值9.5的缓冲液
4、0.1N(0.05M)的EDTA标准液(18.6g/L Na
2EDTA.·2H
2
O)
(B)方法:
1、精确吸取1.0ml样品于250ml锥形瓶中
2、用浓NaOH滴定至沉淀出现
3、加入50ml D I水
4、加入10ml pH值9.5的缓冲液
5、加入3滴PAN指示剂
6、用0.1N的EDTA滴定至由蓝变绿色
(C) 计算:
Cu2+ (g/L) = 64 ×(V. N) EDTA/ Vs
六、产品规格
25升/桶。