grubbs催化剂机理
双环戊二烯开环易位聚合反应用催化剂的研究进展

第49卷第1期上㊀海㊀塑㊀料SHANGHAIPLASTICS㊀Vol.49No.1㊀2021㊀基金项目:上海市科委高新技术领域项目(185****9200)作者简介:时萌珣(1995 )ꎬ男ꎬ在读硕士研究生ꎬ研究方向为聚烯烃催化技术ꎮ通信作者:杨维成(1982 )ꎬ男ꎬ高级工程师ꎻywcjc@163.comꎮ罗㊀勇(1974 )ꎬ男ꎬ教授级高工ꎻluoyongno.1@163.comꎮDOI:10.16777/j.cnki.issn.1009 ̄5993.2021.01.002双环戊二烯开环易位聚合反应用催化剂的研究进展时萌珣1ꎬ㊀刘㊀前1ꎬ㊀刘㊀建1ꎬ㊀段高坤1ꎬ方超立1ꎬ㊀杨维成1ꎬ2ꎬ3ꎬ㊀罗㊀勇1ꎬ2ꎬ3(1.上海化工研究院有限公司ꎬ上海200062ꎻ2.聚烯烃催化技术与高性能材料国家重点实验室ꎬ上海200062ꎻ3.上海市聚烯烃催化技术重点实验室ꎬ上海200062)摘㊀要:聚双环戊二烯(PDCPD)是由双环戊二烯(DCPD)聚合而成的高分子化合物ꎬ作为一种新型高性能树脂ꎬ近年来其市场需求急速发展ꎮ为制备PDCPD材料ꎬ聚合反应中的催化剂是技术关键ꎬ直接决定了产品的性能与制备工艺的经济性ꎮ主要基于催化技术的发展过程ꎬ从催化剂的结构与功能等方面ꎬ介绍了目前DCPD聚合制备PDCPD过程中所采用的主要催化剂的发展现状ꎮ关键词:聚双环戊二烯ꎻ开环易位聚合反应ꎻ催化剂中图分类号:TQ342㊀㊀㊀文献标志码:A㊀㊀㊀文章编号:1009 ̄5993(2021)01 ̄0012 ̄09DevelopmentofCatalystTechnologyinRing ̄openingMetathesisPolymerizationofDicyclopentadieneSHIMengxun1ꎬ㊀LIUQian1ꎬ㊀LIUJian1ꎬ㊀DUANGaokun1ꎬ㊀FANGChaoli1ꎬYANGWeicheng1ꎬ2ꎬ3ꎬ㊀LUOYong1ꎬ2ꎬ3(1.ShanghaiResearchInstituteofChemicalIndustryCo.ꎬLtd.ꎬShanghai200062ꎬChinaꎻ2.StateKeyLaboratoryofPolyolefinsandCatalysisꎬShanghai200062ꎬChinaꎻ3.ShanghaiKeyLaboratoryofCatalysisPolyolefinsꎬShanghai200062ꎬChina)Abstract:Polydicyclopentadiene(PDCPD)ispolymerizedfromdicyclopentadiene(DCPD)ꎬperformingasanexcellentpolymerꎬitsmarketdemandisgrowingrapidlyinrecentyears.InordertopreparePDCPDmaterialsꎬthecatalystsusedinthepolymerizationisthekeytechnologyꎬwhichdirectlydeterminetheperformanceandeconomicsofpreparation.BasedonthedevelopmentprogressofcatalytictechnologyꎬfromthetechnicalaspectsofcatalyststructureandfunctionꎬthemaincatalystsofDCPD spolymerizationareintroducedꎬandreviewedthedevelopmentofcatalysttechnology.Keywords:PDCPDꎻring ̄openingmetathesispolymerizationꎻcatalyst0㊀前言聚双环戊二烯(PDCPD)是一种以双环戊二烯(DCPD)为原料ꎬ经有机金属化合物催化聚合形成的一种具有轻度交联结构的热固性工程树脂ꎬ其最显著特点是力学性能平衡ꎬ兼具刚性和韧性ꎬ具有较高的弯曲强度以及抗冲击强度ꎮ与其他工程塑料相比ꎬ具有优良的综合性能:耐热性优于聚氨酯㊁聚氯乙烯㊁聚乙烯㊁聚丙烯等材料ꎻ尺寸稳定性优于聚氨酯ꎻ抗蠕变性优于尼龙ꎮPDCPD还兼具质轻㊁耐腐蚀㊁耐低温㊁吸水率低㊁涂覆性好等优点ꎮ因此ꎬPDCPD作为新一代高分子材料ꎬ成为当今研究热点ꎬ有望在众多领域成为主导材料ꎮ目前ꎬPDCPD在通信设备㊁电器设备㊁交通设施㊁体育设施㊁铸造配件㊁土木建筑材料中都有广泛应用ꎬ近几年其市场需求也在不断扩大ꎮ1㊀PDCPD简介PDCPD由DCPD通过金属有机化合物催化聚合形成ꎬ具有轻度交联的结构(见图1)ꎮPDCPD聚合反应称为开环易位聚合(ROMP)反应ꎬ反应单元的本质实际是烯烃复分解反应ꎬ反应过程中存在四元环结构中间体(见图2)ꎮ图1㊀PDCPD的聚合和分子结构图2㊀烯烃复分解反应㊀㊀目前公认的聚合反应机理中ꎬ活性中心是金属卡宾(见图3)ꎮ金属卡宾与DCPD中的双键形成含有金属四元环结构的中间体ꎬ随后以易位方式发生裂解ꎬ形成金属卡宾配合物[1]ꎬ这是反应的链引发阶段ꎮ单体继续插入ꎬ增长的金属卡宾与金属四元环不断交替出现ꎬ循环进行[2]ꎬ为链增长阶段ꎮ反应持续进行ꎬ最终形成具有轻度交联的三维网状结构的高分子量聚合物ꎮ图3㊀DCPD聚合反应机理㊀㊀在PDCPD材料的制备过程中ꎬ催化剂是工艺的关键和技术源头ꎬ也是开发材料成型工艺㊁发展PDCPD基复合材料的前提和技术保障[3]ꎮ一方面ꎬ催化剂的活性㊁稳定性直接决定了聚合反应过程和树脂材料的力学性能ꎬ制备成本决定着催化剂的工业应用价值ꎻ另一方面ꎬ催化剂性能更优㊁耐受性更好ꎬ使得PDCPD的功能化应用成为可能ꎮ目前广泛使用的烯烃复分解反应催化体系基本可以分为双组分催化剂和单组分金属卡宾类催化剂ꎮ单组分金属卡宾类催化剂主要为钌卡宾类的催化剂ꎬ其中以Schrock型催化剂㊁Grubbs催化剂㊁Hoveyda ̄Grubbs催化剂最具代表性ꎮ2㊀双组分催化剂最早开始应用于DCPD聚合反应的是双组分催化剂ꎮ双组分催化体系通常由主催化剂和助催化剂组成ꎮ主催化剂为过渡金属的卤化物(如WCl6㊁MoCl5㊁ReCl5㊁RuCl3)或过渡金属氯氧化物(如WOCl4)ꎻ助催化剂为强路易斯酸类物质(如烷基铝及其衍生物㊁SiCl4㊁RMgI㊁苯乙炔等)ꎮ主催化剂在助催化剂的辅助下形成高活性的金属卡宾以催化ROMP反应进行ꎬ主催化剂的质量决定催化所形成的活性物种金属卡宾的质量ꎬ而助催化剂的质量决定金属卡宾的形成速率ꎮ以铝/钼为主催化剂的双组分催化剂研究与工业应用多发生在20世纪80年代ꎮ美国Hercules公司采用Et2AlCl为助催化剂[6]ꎬWCl6/WOCl4为主催化剂[4 ̄5]形成金属卡宾ꎬ以激发催化活性ꎮ同时期ꎬ美国Goodrich公司在专利中采用[(C12H25)3NH]4Mo8O26为主催化剂ꎬEt2AlCl和SiCl4为助催化剂ꎬ成功催化了DCPD的ROMP反应[7 ̄8]ꎮ双组分催化体系比较敏感ꎬ通常在体系中加入醇㊁酚类等物质ꎬ其羟基作为配体与金属中心配位ꎬ减少钨与水㊁氧接触的概率ꎬ形成较稳定的化合物ꎮ这些不同类型配体对钨的电子效应各异ꎬ造成31 第1期㊀时萌珣ꎬ等:双环戊二烯开环易位聚合反应用催化剂的研究进展㊀㊀㊀㊀㊀㊀㊀W Cl键的电子云密度也不同ꎬ与助催化剂反应形成卡宾的速率也有所差别ꎬ因此ꎬ使用不同类型的配体可以得到具有不同活性的催化体系ꎮ此外ꎬDCPD的ROMP反应会快速放热ꎬ即使在室温下反应速率也较快ꎬ往往会产生暴聚现象ꎬ影响聚合反应的稳定性ꎮ为满足反应注射成型工艺的实际操作需求ꎬ常加入路易斯碱㊁胺㊁β ̄二酮㊁醚类作为反应的延缓剂ꎮ催化剂组分中的助催化剂也可调控ꎬ在原先单组分助催化剂的基础上添加其他物质形成双组分助催化剂ꎬ能够更好地控制聚合反应ꎬ如加入Et2AlIꎬ可以控制聚合凝胶时间ꎬ提高PDCPD树脂交联度ꎮ总体而言ꎬ采用双组分催化剂的制备成本较低廉ꎬ体系中形成的钨卡宾㊁钼卡宾对ROMP反应具有较好活性ꎬ至今仍用于PDCPD材料的工业生产ꎮ然而ꎬ此类催化剂缺点明显:一方面催化剂对空气㊁水以及大部分官能团极其敏感ꎬ微量湿气即可影响模具边缘部分催化剂的活性ꎬ造成边缘部分硬化较差ꎻ另一方面其所用的助催化剂(烷基铝)非常不稳定ꎬ遇水爆炸㊁遇氧燃烧ꎬ实际应用十分复杂ꎮ另外ꎬ双组分催化剂难以判断活性中心位置ꎬ研究其机理相对困难ꎬ聚合物的立体结构控制也有一定难度ꎮ3㊀单组分催化剂自1971年法国石油研究院CHAUVINY阐明金属四元环反应机理[1]后ꎬ许多科学家以此机理为基础ꎬ开发出各类高效的单活性中心催化剂ꎮ目前对制备PDCPD用催化剂的研究与应用聚焦在金属卡宾类催化剂上ꎬ即本身结构就含有金属卡宾并可引发聚合的单分子催化剂ꎬ主要代表为Schrock型催化剂和Grubbs催化剂ꎬ以及以Grubbs催化剂为基础改良的Hoveyda ̄Grubbs催化剂ꎮSchrock型催化剂的发现是研究ROMP反应的一个重要里程碑ꎮ通过化学结构上的改变ꎬ可以在更大的范围内调节催化剂活性ꎮ以Grubbs催化剂为代表的钌催化体系更易制备和控制ꎬ活性更强ꎬ多为活性聚合催化剂ꎬ能够催化含有各种不同类型官能团的单体ꎬ在空气和水的介质中表现出良好的稳定性ꎮ3.1㊀钽㊁钨㊁钼体系催化剂钽㊁钨㊁钼体系催化剂的代表即为Schrock型催化剂ꎮSCHROCKRR等[9]合成含钽金属卡宾的配合物[Ta(CHCMe3)3Cl(PMe3)(OCMe3)2]ꎬ其中的钽处于最高氧化价态(+5)ꎮ由于存在叔丁氧基配体ꎬ该配合物的催化活性要高于当时其他类似配合物ꎮ在早期烯烃复分解催化剂的开发研究中ꎬ常以金属钼和钨作为催化中心ꎬSCHROCKRR等[10]基于钼的高氧化态亚烷基配合物ꎬ开发出几种单活性中心钼卡宾化合物ꎬ并得到了通式为[Mo(CHCMe2Ph)(N Ar)(OR)2]的配合物[11 ̄12](见图4)ꎮ㊀㊀图4㊀几种代表性的Schrock型催化剂㊀㊀相比双组分催化剂ꎬSchrock型催化剂引发机理更为清晰ꎮ这些金属卡宾配合物是当时催化活性最高且结构明确的单组分烯烃复分解催化剂ꎮ该类催化剂对大多数不同空间㊁电子效应的底物都具有很高的催化反应活性ꎬ对双键上有单取代㊁二取代或三取代的双键底物都可以得到含相应取代基的双键环合产物ꎬ也是当时唯一能催化四取代双键底物合环的催化剂ꎮ该类催化剂另一显著特点是具有较高的活性和立体选择性ꎬ能形成全同立构和间同立构的聚合物ꎬ且可以催化含有醚㊁酯㊁胺㊁腈㊁膦等极性官能团的单体聚合ꎮ但是ꎬ由于Schrock型催化剂对醛㊁酮质子化的官能团敏感ꎬ以及对空气㊁水ꎬ甚至体系中痕量的杂质敏感ꎬ不易保存ꎻ同时ꎬ其操作必须在惰性气氛41 ㊀㊀㊀㊀㊀㊀㊀上㊀海㊀塑㊀料㊀㊀㊀㊀2021年第49卷㊀下的无水溶剂中进行ꎬ增加了工艺复杂程度ꎬ大大限制了该类催化剂的发展和应用[13]ꎮ此后ꎬ也有科学家开发了新的以金属钼㊁钨为催化中心的催化剂ꎮ2012年ꎬNAYABS等[14]测试了使用[WOCl2 ̄(hpap)]的顺式和反式异构体的混合物催化环烯烃(如四环十二烯和四环十二烯甲酸甲酯)的ROMP反应ꎬ使用三烷基铝作为促进剂ꎮ该催化剂被证明具有很高的活性(见图5)ꎬ其对空气非常敏感ꎬ必须在无氧环境下进行操作ꎬ增加了工艺的复杂性ꎮ图5㊀NAYABS等在2012年测试的催化剂㊀㊀LEHTONENA等[15]研究了具有螯合酚盐(L)的亚胺钨(VI)配合物(见图6)ꎮ[W(NPh)Cl3(L)]类型的VI配合物可以被乙基溴化镁活化ꎬ催化降冰片烯衍生物(如2 ̄降冰片烯㊁5 ̄乙烯基 ̄2 ̄降冰片烯㊁DCPD)的ROMP反应ꎮ当用乙基溴化镁处理时ꎬ这些化合物形成活性催化剂ꎮ聚合反应可以在环境气氛下进行ꎬ不需要复杂的惰性气氛技术ꎬ反应产生具有高顺式含量的聚合物ꎮ㊀㊀图6㊀LEHTONENA等在2008年研究的催化剂㊀㊀2020年ꎬBENEDIKTERMJ等[16]合成并测试了大量以VI族金属元素为金属中心的催化剂(见图7)ꎬ其中大部分都具有高效率的催化性能ꎬ但大多数都对许多官能团十分敏感ꎬ聚合体系中的空气㊁水㊁醇等物质也会使其失效ꎬ仅有少数几种(图7中两者为代表)可以应用ꎮ这几种催化剂的稳定性和官能团耐受性已媲美目前带氮杂环卡宾(NHC)配体的钌基催化剂ꎮ㊀㊀图7㊀BENEDIKTERMJ等在2020年研究的催化剂3.2㊀钌体系催化剂自从NGUYENST等[17]在1992年提出了第一种结构明确的钌卡宾配体催化剂以来ꎬ以钌为金属中心㊁卡宾类结构为配体的催化剂因其高活性㊁高效率的催化性能和易于合成的特性ꎬ使烯烃复分解反应催化剂的研究主要集中在了钌体系催化剂上ꎮGrubbs催化剂是以钌为金属中心的金属卡宾配合物ꎬ通式为[Ru(CHR)Cl2(L)(L )](见图8)ꎮ为了提高催化剂的反应活性ꎬGRUBBS等将分子式为[RuCl2(PPh3)2(CH CHCHPh)]的配合物结构中与磷相连的苯基(Ph)换成环己基(Cy)ꎬ在1995年提出了Grubbs第一代催化剂[18]ꎬ见图8(a)ꎮ结果提高了催化反应的活性ꎬ大大加快了反应速率[19 ̄20]ꎮ(a)第一代催化剂㊀(b)第二代催化剂图8㊀Grubbs催化剂㊀㊀Grubbs第一代催化剂合成步骤简单㊁结构稳定不易分解㊁催化活性较高ꎬ且具有很好的官能团兼容性[21]ꎬ在质子溶剂中也很稳定[22]ꎮ通常情况下ꎬ对酰胺类底物的环化产率比较高ꎬ更明显的优点是该催化剂不受空气㊁水以及体系中杂质的影响ꎬ因此扩大了应用范围ꎮ但是ꎬ该催化剂不适用于含胺基的底物ꎬ胺基的存在会使其失去活性ꎮ为进一步改进催化剂性能ꎬ1999年GRUBBS等在研究催化反应机理时发现ꎬ在催化反应的引发阶段存在膦配体与金属中心解离的过程ꎬ进而产生51 第1期㊀时萌珣ꎬ等:双环戊二烯开环易位聚合反应用催化剂的研究进展㊀㊀㊀㊀㊀㊀㊀一个具有催化活性的中间体ꎮ如果能有效地加快膦配体与金属中心的解离速率ꎬ则该催化剂的催化效率就有可能提高ꎮSCHOLLM等[23]经过进一步研究得出结论:催化反应的发生需要钌卡宾配合物分子中的一个膦配体解离生成活泼的钌中间体ꎮ为此ꎬGRUBBS研究团队将原有结构中的一个膦配体换成具有大空间位阻的NHC配体[24]ꎬ得到了Grubbs第二代催化剂[25]ꎬ见图8(b)ꎮ该催化剂延续了Grubbs第一代催化剂的特点ꎬ稳定㊁易制备ꎬ露置在空气中储存数星期都不会分解ꎮ同时反应条件温和ꎬ具备较高的催化活性ꎬ反应时间更短ꎬ催化剂用量更少[26]ꎮ这一烯烃复分解反应催化剂因其广泛的有机官能团适用性和在空气中的稳定性ꎬ受到有机化学家的青睐ꎮ㊀㊀Grubbs催化剂热稳定性较差ꎬ在较高的温度下易发生分解ꎮ1999年KINGSBURYJS等[27]在Grubbs催化剂的基础上开发得到了异丙氧基螯合的Hoveyda ̄Grubbs第一代催化剂ꎬ见图9(a)ꎮ随后又在2000年进一步开发得到了不含膦配体的Hoveyda ̄Grubbs第二代催化剂[28]ꎬ见图9(b)ꎮ其中在分子中引入具有较大体积的亲核性异丙氧螯络合物配体ꎬ提高了催化剂的热稳定性ꎬ并且在催化反应时有较高的引发速率ꎮ在催化反应中ꎬ尤其是Hoveyda ̄Grubbs第二代催化剂ꎬ在室温条件下反应不到2h就可以获得88%的收率[29]ꎮ(a)第一代催化剂(b)第二代催化剂图9㊀Hoveyda ̄Grubbs催化剂㊀㊀第一代和第二代的Hoveyda ̄Grubbs催化剂分别由相应的Grubbs催化剂衍生ꎬ原Grubbs催化剂中一个三环己基膦基团被苯环邻位的异丙氧基所替代ꎮHoveyda ̄Grubbs第一代催化剂适用于末端烯烃的关环复分解反应ꎮHoveyda ̄Grubbs第二代催化剂适用于缺电子烯烃的关环㊁开环和交叉复分解反应ꎮ4㊀其他钌体系催化剂由于钌体系催化剂相较于钼㊁钨类催化剂而言ꎬ对大量有机官能团㊁水分和氧气的耐受性能更佳ꎬ所以近十几年来对于烯烃复分解类反应催化剂的研究聚焦在钌卡宾配合物类催化剂上ꎮDCPD的ROMP反应会快速放热ꎬ但Grubbs催化剂㊁Hoveyda ̄Grubbs催化剂中的钌卡宾配合物都具有较高的催化活性ꎬ聚合反应在数分钟内完成ꎬ即使在室温下反应速率也较快ꎮ这使得反应不易控制ꎬ容易产生暴聚现象ꎬ影响聚合反应的稳定性ꎬ造成产物的收率降低㊁杂质增加等不良后果ꎬ导致聚合物性能降低ꎮ为此ꎬ在反应时不得不加入缓聚剂ꎬ降低反应速率以提高反应的稳定性ꎬ但这样又增加了聚合工艺的复杂性ꎬ引入了杂质ꎬ使聚合物的后处理难度增加ꎮ因此ꎬ近十几年来不少科学家研究在钌卡宾配合物上加上特定配体ꎬ延缓聚合反应的发生ꎬ或者使聚合反应在某特定条件下才能引发ꎬ提高反应的稳定性ꎮ为增强Grubbs催化剂的稳定性ꎬSAMECJSM等[30]在2007年对其先前开发的催化剂进行了配体上的改良ꎬ在Grubbs第二代催化剂的基础上ꎬ增加了一个半稳定性的双齿配体(见图10)ꎬ抑制了催化剂在反应过程中的分解ꎬ使副反应更少发生ꎬ同时其催化活性能够在反应进行过程中随着温度的升高逐渐引发ꎬ减少暴聚现象ꎮ另外ꎬ该催化剂在合成过程中还避免了以Grubbs催化剂为基础进行改进时常会使用到的铊盐ꎮ图10㊀GRUBBS等在2007年开发的催化剂61 ㊀㊀㊀㊀㊀㊀㊀上㊀海㊀塑㊀料㊀㊀㊀㊀2021年第49卷㊀㊀㊀但是ꎬ该催化剂的不足之处在于ꎬ在同样条件下的催化活性较Grubbs催化剂略有降低ꎬ限制了进一步的应用ꎮ有科学家发现ꎬ当催化剂结构存在含有OңRu㊁NңRu等配位键的双齿螯合配体时ꎬ催化剂活性引发的过程与OңRu㊁NңRu等配位键的断裂有关[31]ꎮ随着这些配位键的断裂ꎬ双齿螯合配体变为单齿非螯合配体ꎬ催化剂结构中的钌卡宾部分暴露出来ꎬ催化剂得以显现出活性ꎬ可以进行下一步的反应(见图11)ꎮ图11㊀Hoveyda ̄Grubbs第二代催化剂中的OңRu配位键的断裂㊀㊀近十几年来ꎬ众多研究者着眼于催化剂活性引发的机理ꎬ基于Grubbs催化剂ꎬ开发出许多新的催化剂ꎬ其中大部分都使用了多齿螯合配体ꎬ增强了催化剂在反应过程中的性能ꎬ还开发了一些新的反应功能ꎮ该类钌卡宾配合物在室温下的反应中不活泼ꎬ但随后可被一些引发条件ꎬ如热㊁光㊁酸㊁化学活化㊁超声波等活化ꎮ在钌金属中心上使用螯合卡宾配体是合成高性能催化剂的较有希望的方法ꎮ这种螯合配体是双齿的ꎬ在室温下稳定了催化剂的静息状态ꎬ而在一定条件下会释放出一个配位位点ꎬ使得聚合反应可以进行ꎮ从使用双齿螯合配体的思路出发ꎬSZADKOWSKAA等[32]在2009年发表的研究给出了具有代表性的解决方法ꎮ他们在Grubbs第二代催化剂的基础上进行了改进ꎬ制备了一系列新型催化剂ꎬ使钌卡宾配合物具有了空间位阻更大的卡宾螯合配体ꎬ在催化剂保持活性的同时使其稳定性大大提高(见图12)ꎮ图12㊀SZADKOWSKAA等在2009年开发的催化剂㊀㊀图12中合成的催化剂ꎬ在催化降冰片烯衍生物的ROMP反应时ꎬ当反应温度低于80ħ时ꎬ聚合反应几乎不发生ꎬ而当反应温度在120ħ左右时ꎬ又能获得较高的催化效率[32]ꎬ实现了反应的温度控制ꎮ㊀㊀但是ꎬ该催化剂由于合成路线较为复杂ꎬ产率低下ꎬ不利于进一步广泛应用ꎮ之后ꎬSZWACZKOK等[33]继续在2017年发表71 第1期㊀时萌珣ꎬ等:双环戊二烯开环易位聚合反应用催化剂的研究进展㊀㊀㊀㊀㊀㊀㊀了进一步的研究成果ꎬ合成㊁测试并挑选了一批表现优秀的催化剂(见图13)ꎮ其中ꎬ较有应用价值的是含有偶氮结构配体的催化剂ꎬ其催化活性引发温度在100ħꎬ其催化活性也略有提高ꎮ㊀㊀㊀图13㊀SZWACZKOK等在2017年开发的催化剂㊀㊀BEN ̄ASULYA等[34]也对Hoveyda ̄Grubbs第二代催化剂进行了改良ꎬ在2008年合成了一种新型的催化剂(见图14)ꎬ将Hoveyda ̄Grubbs第二代催化剂结构中的O替换成了Sꎬ发现该催化剂在催化聚合反应中表现出了温控作用ꎮ催化聚合反应在室温下几乎不发生ꎬ而反应在升温至80ħ时催化剂的活性开始显现ꎮ图14㊀BEN ̄ASULYA等在2008年开发的催化剂㊀㊀LEXERC等[35]又在2011年测试了一种带有螯合结构的新型催化剂ꎬ以Hoveyda ̄Grubbs催化剂为基础开发ꎬ该催化剂中的三苯基膦(PPh3)配体的其中一个苯基和连接钌的苯乙基卡宾合二为一ꎬ形成了一个双齿螯合配体(见图15)ꎮ使得该催化剂具有了温控反应特性ꎬ可以在42ħ以下保持稳定的催化活性ꎬ而在60ħ以上激发出高催化活性ꎮ图15㊀LEXERC等在2011年开发的催化剂㊀㊀ÖZTÜRKABÖ等[36]尝试了另一种反应控制思路 酸引发ꎮ他们在2015年发布了一种新型的带有咪唑官能化的席夫碱配体的钌 ̄茚并亚烷基催化剂(见图16)ꎬ用于水乳液中的ROMPꎬ可以通过添加酸来活化并催化降冰片烯结构的ROMPꎮ通过改变酸/钌比ꎬ可以自由控制水乳液中ROMP的 81 ㊀㊀㊀㊀㊀㊀㊀上㊀海㊀塑㊀料㊀㊀㊀㊀2021年第49卷㊀开关ꎬ进而可以控制乳液聚合物的分子质量ꎮ图16㊀ÖZTÜRKABÖ等在2015年开发的催化剂5㊀结语依据PDCPD聚合反应催化剂的发展进程ꎬ从催化效率与稳定性能等角度出发ꎬ主要综述了应用在DPCD聚合反应中的几大体系催化剂ꎬ以及最新的研究进展ꎮ结果表明:将带有螯合配体的钌卡宾类催化剂应用在PDCPD聚合反应是近年来的研究热点ꎬ研究者们在继续探索聚合反应中催化剂生效机理的同时ꎬ开发出了许多高性能的催化剂ꎮ引入温控引发㊁酸引发等新功能也是备受关注的领域ꎬ有不少研究者为了使催化聚合反应更可控开发出更多的新型功能性催化剂ꎮ但是ꎬ这些新型催化剂因其合成困难㊁产量低㊁价格昂贵㊁经济性差等缺点ꎬ仍然存在工业化障碍ꎮ有理由相信ꎬ未来将会有越来越多的研究关注于DPCD聚合反应中更高性能的新型聚合反应催化剂ꎮ参考文献:[1]㊀CHAUVINYꎬHERISSONJL.Catalysisofolefintransformationsbytungstencomplexes.II.Telomeriza ̄tionofcyclicolefinsinthepresenceofacyclicolefins[J].DieMakromoleKulareCharieꎬ1971ꎬ141(1):161 ̄167.[2]㊀PATRICIOERꎬWARRENEP.Mechanisticstudieson14 ̄electronruthenacyclobutanes:Degenerateex ̄changewithfreeethylene[J].JournaloftheAmericanChemicalSocietyꎬ2007ꎬ129(6):1698 ̄1704. [3]㊀乔新峰ꎬ杨维成ꎬ付宏伟ꎬ等.聚双环戊二烯反应注射成型的研究进展[J].上海塑料ꎬ2017(4):9 ̄14.[4]㊀NELSONLL.Thermallyactivatedtwocomponentme ̄tathesiscatalystsystem:US4727125[P].1988 ̄02 ̄23. [5]㊀NELSONLL.Ratemoderatedtwocomponentmetathe ̄siscatalystsystem:US4897456[P].1990 ̄01 ̄30. [6]㊀KLOSIEWICZDW.Cycloolefinpolymerizationcatalystcomposition:US4568660[P].1986 ̄02 ̄04. [7]㊀LANEPCJr.ꎬFallsCꎬetal.Liquiddicyclopenta ̄dienefeedstockforbulkpolymerization:US4906797[P].1990 ̄03 ̄06.[8]㊀NGUYENTTꎬDELW.Methodformakingadicyclo ̄pentadienecross ̄linkedpolymerandtheproductthere ̄of:US4808635[P].1989 ̄02 ̄28.[9]㊀SCHROCKRRꎬMEAKINP.Pentamethylcomplexesofniobiumandtantalum[J].JournaloftheAmericanChemicalSocietyꎬ1974ꎬ96(16):5288 ̄5290. [10]㊀SCHROCKRRꎬMURDZEKJSꎬBAZANGCꎬetal.Synthesisofmolybdenumimidoalkylidenecomplexesandsomereactionsinvolvingacyclicolefins[J].Jour ̄naloftheAmericanChemicalSocietyꎬ1990ꎬ112(10):3875 ̄3886.[11]㊀BAZANGCꎬKHOSRAVIEꎬSCHROCKRRꎬetal.Livingring ̄openingmetathesispolymerizationof2ꎬ3 ̄difunctionalizednorbornadienesbyMd:CHBu ̄tert(:NC6H3Pr ̄1502 ̄2ꎬ6)(OButert)2[J].JournaloftheAmericanChemicalSocietyꎬ1990ꎬ112(23):8378 ̄8387.[12]㊀BAZANGCꎬOSKAMJHꎬSCHROCKRRꎬetal.Livingring ̄openingmetathesispolymerizationof2ꎬ3 ̄difunctionalized7 ̄oxanorbornenesand7 ̄oxanorbornadi ̄enes[J].JournaloftheAmericanChemicalSocietyꎬ1991ꎬ113(18):6899 ̄6907.[13]㊀朱杰ꎬ张学景ꎬ邹永.关环复分解(RCM)及其催化剂研究进展[J].有机化学ꎬ2004ꎬ24(2):127 ̄139.[14]㊀NAYABSꎬPARKWꎬWOOHYꎬetal.SynthesisandcharacterizationofnoveltungstencomplexesandtheiractivityintheROMPofcyclicolefins[J].Poly ̄hedronꎬ2012(42):102 ̄109.[15]㊀LEHTONENAꎬBALCARBHꎬSILLANPÄÄDRꎬetal.SynthesisandROMPactivityofaminophenol ̄substitutedtungsten(VI)andmolybdenum(VI)com ̄plexes[J].JournalofOrganometallicChemistry.2008ꎬ693(7):1171 ̄1176.[16]㊀BENEDIKTERMJꎬZIEGLERFꎬGROOSJꎬetal.Group6metalalkylideneandalkylidyneN ̄heterocycliccarbenecomplexesforolefinandalkynemetathesis[J].CoordinationChemistryReviewsꎬ2020ꎬ415(213315):1 ̄31.[17]㊀NGUYENSTꎬGRUBBSRHꎬZILLERJW.Synthe ̄sesandactivitiesofnewsingle ̄componentꎬruthenium ̄91第1期㊀时萌珣ꎬ等:双环戊二烯开环易位聚合反应用催化剂的研究进展㊀㊀㊀㊀㊀㊀㊀basedolefinmetathesiscatalysts[J].JournaloftheA ̄mericanChemicalSocietyꎬ1993ꎬ115(21):9858 ̄9859.[18]㊀SCHWABPꎬGRUBBSRHꎬZILLERJW.SynthesisandapplicationsofRuCl2(=CHR')(PR3)2:Thein ̄fluenceofthealkylidenemoietyonmetathesisactivity[J].JournaloftheAmericanChemicalSocietyꎬ1996ꎬ118(1):100 ̄110.[19]㊀TRNKATMꎬGRUBBSRH.ThedevelopmentofL2X2Ru=CHRolefinmetathesiscatalysts:anorgano ̄metallicsuccessstory[J].AccountsofChemicalRe ̄searchꎬ2001ꎬ34(1):18 ̄29.[20]㊀SCHWABPꎬZILLERJWꎬGRUBBSRHꎬetal.Aseriesofwell ̄definedmetathesiscatalysts ̄synthesisof[RuCl2(CHR)(PR3)2]anditsreactions[J].Ange ̄wandteChemieInternationalEditionꎬ1995ꎬ34(18):2039 ̄2041.[21]㊀ROMEROPEꎬPIERSWEꎬMCDONALDR.Rapidlyinitiatingrutheniumolefin ̄metathesiscatalysts[J].AngewandteChemieInternationalEdition.2004ꎬ43(45):6161 ̄6165.[22]㊀BAIBICHIMꎬKERNC.Reactivityoftungsten ̄arylo ̄xideswithhydrosilanecocatalystsinolefinmetathesis[J].JournaloftheBrazilianChemicalSocietyꎬ2002ꎬ13(1):43 ̄46.[23]㊀SCHOLLMꎬDINGSꎬGRUBBSRHꎬetal.Synthesisandactivityofanewgenerationofruthenium ̄basedo ̄lefinmetathesiscatalystscoordinatedwith1ꎬ3 ̄dimes ̄ityl ̄4ꎬ5 ̄dihydroimidazol ̄2 ̄ylideneLigands[J].Or ̄ganicLettersꎬ1999ꎬ1(6):953 ̄956.[24]㊀马玉国.烯烃复分解反应 2005年诺贝尔奖简介[J].大学化学ꎬ2006ꎬ24(1):1 ̄7.[25]㊀FUGCꎬNGUYENSTꎬGRUBBSRH.Catalyticring ̄closingmetathesisoffunctionalizeddienesbyaru ̄theniumcarbenecomplex[J].JournaloftheAmericanChemicalSocietyꎬ1993ꎬ115(21):9856 ̄9857. [26]㊀GILBERTBCꎬKALZWꎬLINDSAYCIꎬetal.Initi ̄ationofradicalcyclisationreactionsusingdimanganesedecacarbonyl:Aflexibleapproachtopreparing5 ̄mem ̄beredrings[J].JournaloftheChemicalSocietyꎬ2000ꎬ8(1):1187 ̄1194.[27]㊀KINGSBURYJSꎬHARRITYJPAꎬHOVEYDAAHꎬetal.ArecyclableRu ̄basedmetathesiscatalyst[J].JournaloftheAmericanChemicalSocietyꎬ1999ꎬ121(4):791 ̄799.[28]㊀GARBERSBꎬKINGSBURYJSꎬHOVEYDAAHꎬetal.EfficientandrecyclablemonomericanddendriticRu ̄basedmetathesiscatalysts[J].JournaloftheA ̄mericanChemicalSocietyꎬ2000ꎬ122(34):8168 ̄8179.[29]㊀RANDLSꎬGESSLERSꎬBLECHERTSꎬetal.HighlyselectivecrossmetathesiswithacrylonitrileusingaphosphinefreeRu ̄complex[J].Synlettꎬ2001(3):430 ̄432.[30]㊀SAMECJSMꎬGRUBBSRH.RutheniumcarbenecomplexesbearingananionicCarboxylatechelatedtoahemilabileligand[J].ChemistryEuropeJournalꎬ2008ꎬ14(9):2686 ̄2692.[31]㊀DENKKꎬFRIDGENJꎬHERRMANNWA.N ̄hetero ̄cycliccarbenesꎬpart33.[1]combiningstableNHCandchelatingpyridinyl ̄alcoholatoligands:Arutheniumcatalystforapplicationsatelevatedtemperatures[J].AdvancedSynthesis&Catalysisꎬ2010ꎬ344(6 ̄7):666 ̄670.[32]㊀SZADKOWSKAAꎬGSTREINXꎬGRELAKꎬetal.Latentthermo ̄switchableolefinmetathesisinitiatorsbearingapyridyl ̄functionalizedchelatingcarbene:in ̄fluenceoftheleavinggroup'srigidityonthecatalyst'sperformance[J].Organometallicsꎬ2010ꎬ29(1):117 ̄124.[33]㊀SZWACZKOKꎬCZELUSNIAKIꎬGRELAK.Apar ̄tiallyserendipitousdiscoveryofthermo ̄switchableru ̄theniumolefinmetathesisinitiatorthatseemtobewellsuitedforROMPofmonomersbearingvinylpendantgroups[J].JournalofOrganometallicChemistryꎬ2017ꎬ847(1):146 ̄153.[34]㊀BEN ̄ASULYAꎬTZUREꎬLEMCOFFNGꎬetal.AthermallyswitchablelatentrutheniumolefinmetathesisCatalyst[J].Organometallicsꎬ2008ꎬ27(5):811 ̄813.[35]㊀LEXERCꎬLEMCOFFNGꎬSLUGOVCCꎬetal.Ole ̄finmetathesiscatalystbearingachelatingphosphineligand[J].JournalofOrganometallicChemistryꎬ2011ꎬ696(11 ̄12):2466 ̄2470.[36]㊀ÖZTÜRKABÖꎬSꎬEHITO㊅GLUASKꎬMEIERMAR.Alatentandcontrollableruthenium ̄indenylidenecata ̄lystforemulsionROMPinwater[J].EuropeanPoly ̄merJournalꎬ2015ꎬ62(1):116 ̄123.(收稿日期:2020 ̄04 ̄21)02 ㊀㊀㊀㊀㊀㊀㊀上㊀海㊀塑㊀料㊀㊀㊀㊀2021年第49卷㊀。
烯烃复分解反应

RCM的研究进展
RCM 早期的发展 W 系催化剂
WCl6+Me4Sn 低收率
适用含S,Si,P,Sn底物,二烯丙基 成五元环反应 不适用双键二取代和烯丙基位 有取代基底物
适用5,6元环的合成
+ PbEt4
缺点:需要高温完成从5 到卡宾中间体的转变,
Fu,G.C. J.Am.Chem.Soc.1993,115,3800 Barrett,A.G. Chem. Commun.1996,2231
RCM的研究进展
Schrock 催化剂的应用 氮杂环的合成
适用于三级胺和酰胺, 二级胺稍差
不适用于一级胺和, 或,不饱和胺
含高取代双键的底物 反应可进行
RCM的研究进展
Grubbs催化剂的应用 药物合成
Biswas,K. J.Am.Chem.Soc, 2002,124,9825
RCM的研究进展
固载型催化剂
目的: 保持匀相催化剂 的高活性,高选择性;易 于分离;循环使用;产物 含低金属离子和污染物 浓度
方法: 表面金属有机化 学(Surface organometallic chenistry,SOMC)
耐hedron lett. 1980,21,1715; Couturier,J.L. Angew.Chem.Int.Ed.1992,31,268;Nugent,W.A.J.Am.Chem.Soc.1995,117,8992
RCM的研究进展
Schrock 催化剂
优点: 对广泛的底物有较高的活性, 底物双键可以单,二和三取代,产物 可以是二,三和四取代 缺点: 对空气,H2O,合溶剂中的痕 量杂质敏感,不易储存
基础有机化学反应总结
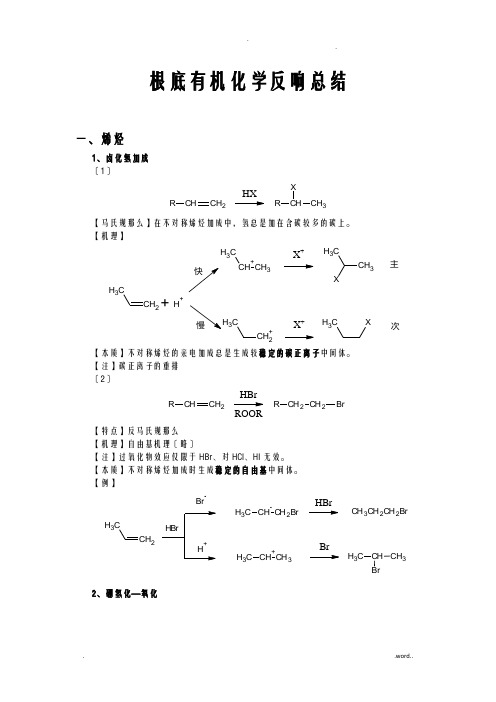
根底有机化学反响总结一、烯烃1、卤化氢加成 〔1〕CHCH 2RHXCHCH 3RX【马氏规那么】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】CH 2CH 3CH +CH 3CH 3X +CH 3CH 3X+H +CH 2+C3X +CH 3X主次【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。
【注】碳正离子的重排 〔2〕CHCH 2RCH 2CH 2R BrHBrROOR【特点】反马氏规那么【机理】自由基机理〔略〕【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】CH 2CH3BrCH CH 2BrC H 3CH +CH 3C H 3HBrBrCH 3CH 2CH 2BrCH BrCH 3C H 32、硼氢化—氧化CHCH 2R CH 2CH 2R OH1)B 2H 62)H 2O 2/OH-【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】CH2CH 33H 323H 32CH CH 2CH 3HBH 2CH CH=CH (CH 3CH 2CH 2)3B-H 3CH 2CH 2C22CH 3CH 2B OCH 2CH 2CH 3H 3CH 2CH 2CCH 2CH 2CH 3+OH -OHB-OC H 2CH 2CH 3CH 2CH 2CH 3H 3CH 2CH 2OBOC H 2CH 2CH 3CH 2CH 2CH 3OC H 2CH 2CH 3HOO -B(OCH 2CH 2CH 3)3B(OCH 2CH 2CH 3)3+3NaOH 3NaOH3HOC H 2CH 2CH 33+Na 3BO 32【例】CH 31)BH 32)H 2O 2/OH-CH 3HH OH3、X 2加成C CBr /CCl CC BrBr【机理】CC CC Br BrC CBr +CC Br OH 2+-H +CC Br OH【注】通过机理可以看出,反响先形成三元环的溴鎓正离子,然后亲和试剂进攻从反面进攻,不难看出是反式加成。
硕士_均相催化原理与应用-4

氢硅烷化反应
The addition of a silicon compound such as R3SiH to alkene functionality, as shown by reaction 1, is used widely in silicone polymer manufacture and is called the hydrosilylation reaction.
24
加氢反应
The industrial importance of homogeneous hydrogenation lies mainly in the fact that by using a chiral catalyst hydrogenated chiral products could be obtained from prochiral alkenes
45
谢Hale Waihona Puke 大家!欢迎提出问题和任何建议!
houzhenshan@ Tel: 64251686(O)
46
3
催化剂的定义
• 1919年 Ostwald (奥斯瓦尔德):“催化剂能影响化学反应 的速度,但它本身并不因化学反应的结果而消耗,它亦不 会改变反应的最终的热力学平衡”。 • 1976年IUPAC(国际纯粹及应用化学协会):“催化作用 是一种化学作用,是靠用量极少而本身不被消耗的一种叫 做催化剂的外加物质来加速化学反应的现象”。 • 1994年国家自然科学基金委员会:“催化是加速反应速度、 控制反应方向或产物构成,而不影响化学平衡的一类作用。 起这种作用的物质称为催化剂,它不在主反应的化学计量 式中反映出来,即在反应中不被消耗。”
氢甲酰化过程
OXO Process(Ruhrchemie AG discovered in 1938):
[正式版]烯烃的复分解反应——机理及实例ppt资料
![[正式版]烯烃的复分解反应——机理及实例ppt资料](https://img.taocdn.com/s3/m/3e489060a0116c175e0e487a.png)
开环复分解聚合
在这个反应中,金属卡宾化合物(metal methylene)作为催化 剂,反应是可逆的,如果将生成的乙烯移出反应,可使之 反应完全,其机理如下:
二、催化剂的结构
• 某些连有强给电子取代基的卡宾非常稳定。例 如下列卡宾能分离出来。
Hale Waihona Puke NNNN
N
N
在所给出的卡宾结构中,具有三个取代基的苯环保护卡宾, 避免亲核试剂和氧的进攻;同时由于氮原子强给电子的能力, 使得该卡宾不仅不受亲核试剂的进攻,反而具有亲核性能, 因而能像磷那样,作为一种很好的配体,与过渡金属形成配合物。
为大家表演烯烃复分解反应的含
义。最初两位男士是一对舞伴,
两位女士是一对舞伴,在“加催
化剂”的喊声中,他们交叉换位,
转换为两对男女舞伴,在场记者
随即发出了笑声。
金属卡宾络合物催化的烯烃复分解反应 :
金属卡宾化合物
+
[M]
R1
R1
R1 +
R1
生成的烯烃是Z形和E形 的混合物
双键在碳链的末端,生成一乙烯分子和另两端结合的大分子
2. 交错复分解
他亲自走出讲台,邀请身边的皇家科学院教授和 生成的烯烃是Z形和E形 金属卡宾络合物催化的烯烃复分解反应 : 瑞典皇家科学院2005年10月5日宣布,将2005年诺贝尔化学奖授予法国化学家伊夫·肖万(Yves ChauvinFrance )、 化学家罗伯特·格 拉布(Robert H. 避免亲核试剂和氧的进攻;
最新催化剂
三、反应历程
R1 [M]
R1
M
R1
R1
R1 M R1
[M]
R1
R1
Chauvin catalytic cycle
烯烃复分解反应.doc

烯烃复分解反应(英语:Olefin metathesis)涉及金属催化剂存在下烯烃双键的重组,[1]自发现以来便在医药和聚合物工业中有了广泛应用。
相对于其他反应,该反应副产物及废物排放少,更加环保。
2005年的诺贝尔化学奖颁给了化学家伊夫·肖万、罗伯特·格拉布和理查德·施罗克,以表彰他们在烯烃复分解反应研究和应用方面所做出的卓越贡献。
[2]烯烃复分解反应由含镍、钨、钌和钼的过渡金属卡宾配合物催化,反应中烯烃双键断裂重组生成新的烯烃,通式如下:反应机理根据伍德沃德-霍夫曼规则,环加成反应是对称禁阻的,活化能很高。
20世纪70年代时,Hérison广泛接受的反应机制。
[5]其中,首先发生烯烃双键与金属卡宾配合物的[2+2]环加成反应,生成金属杂环丁烷衍生物中间体。
然后该中间体经由逆环加成反应,既可得到反应物,也可得到新的烯烃和卡宾配合物。
新的金属卡宾再与另一个烯烃发生类似的反应,最后生成另一个新的烯烃,并再生原金属卡宾。
金属催化剂d轨道与烯烃的相互作用降低了活化能,使烯烃复分解反应在适宜温度下就可发生,摆脱了以前多催化组分以及强路易斯酸性的反应条件。
复分解反应复分解反应又可分为以下几种重要类型:∙交叉复分解反应∙关环复分解反应∙烯炔复分解反应∙开环复分解反应∙开环复分解聚合反应∙非环二烯复分解反应∙炔烃复分解反应∙烷烃复分解反应∙烯烃复分解反应与大多数有机金属反应类似的是,复分解反应生成热力学控制的产物。
也就是说,最终的产物比例由产物能量高低决定,符合玻尔兹曼分布。
复分解反应的驱动力往往不相同:∙ 烯烃复分解反应和炔烃复分解反应—乙烯/乙炔的生成增加了反应熵,推动了反应发生;∙ 烯炔复分解反应—没有以上条件,在热力学上是不利的,除非还伴随有特定的开环或关环反应;∙ 开环复分解反应—原料常为有张力的烯烃如降冰片烯,环的打开消除了张力,推动了反应发生;∙关环复分解反应—生成了能量上有利的五六元环,反应中通常有乙烯生成。
加成和消除反应PPT课件(高等有机化学)

R2C CHR H X R2CCH2R X 有利
R2C
CHR H
X
X R2CHCHR
不利
R2CCH2R X
R2CHCHR X
当供电子基团与烯烃相连时:
Y
H
Cl C C H + Y
Cl C C H 或 Cl C C H
HH
HH (I)
YH (II)
形成的碳正离子和卤素的未共享电子对共轭,使
体系能量降低。
(3)共轭烯烃的加成反应
❖ 共轭烯烃的亲电加成也是朝形成相对稳定的中间体方向进行。
第一步反应,亲电试剂的亲电部分总是加到共轭链的一个末 端碳上,而不会加到共轭链的中间碳上。如1,3-丁二烯与 HCl反应:
CH2 CH CH CH2
CH2 CH CH
H2C CH CH CH2 + H+ 慢
H
(1)
× H2C CH CH CH2
(2) + Cl
H
(3)
快 CH2 CH CH CH2 + CH2 CH CH CH2
Cl H (4) Cl
H (5)
CH2 H (2)
❖ 第一步慢反应是H+或HCl分子的δ+ 端进攻共轭体系的末端碳 原子生成(2),而不是进攻共轭体系中间碳原子生成(3)。 因为碳正离子(2)比(3)稳定容易生成。第二步快反应是 碳正离子(2)与
影响反应机理的因素,消除反应的定向; E2反应中的立体化学(重点和难点)。 4、了解其它消除反应
二、教学课时: 2
一、亲电加成反应机理
(1)环正离子中间体(反式加成)
E
+
C=C
+ E+Y -
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
grubbs催化剂机理
Grubbs催化剂是一种重要的烯烃Metathesis催化剂,被广泛应用于有机合成与材料科学领域。
该催化剂的机理主要涉及到金属卡宾中间体的形成和分解过程。
在反应开始时,Grubbs催化剂通过与底物中的烯烃发生配位作用,形成一个活性的配合物。
然后,该配合物通过一个交换反应,将金属卡宾中间体生成,该过程涉及到金属与底物中的π键发生断裂和形成的反应。
生成的金属卡宾中间体可以与另一个底物中的烯烃发生反应,产生一种新的烯烃产物,并释放出Grubbs催化剂。
这个过程
被称为烯烃Metathesis反应。
在反应过程中,Grubbs催化剂的活性位点是一种高度活性的金
属卡宾中间体。
这个金属卡宾中间体可以通过多种方式分解。
最常见的是通过氧化还原反应,将其还原回到活性的Grubbs催化剂状态。
这个过程需要引入外部还原剂,比如甲醇或异丙醇等。
此外,金属卡宾中间体也可以被其他分子中的亲核性物质攻击,导致分解反应发生。
总之,Grubbs催化剂机理涉及到金属卡宾中间体的形成和分解
过程。
这个机理为烯烃Metathesis反应提供了一个理论基础,也可
以用于设计和优化新的催化剂,以满足不同的研究需求。
- 1 -。