药物分析 第4章 药物的检查
第四章 药物定量分析与分析方法验证

1、含卤素有机药物
含卤素有机药物是指分子结构中所含卤素直接与芳环
如果测得理论板数低于规定的最小理论板数, 应改变色谱柱的某些条件(如柱长、载体性能、色 谱柱填充的优劣等),使理论板数达到要求。
(2)分离度(R)
在定量分析时,要求定量峰与其他峰 或内标峰之间有较好的分离度。除另有规定 外,分离度应大于1.5。
分离度(R)的计算公式见下式:
R 2 t R2 t R1
含量(CX)= f
AX AS / CS
式中,AX为供试品峰面积或峰高;CX为供
试品的浓度 。
丙酸睾酮含量测定采用高效液相法:取本品 对照品适量,精密称定,加甲醇定量稀释成 每lmL中约含1mg的溶液。精密量取该溶液 和内标溶液(1.6mg/ml苯丙酸诺龙甲醇溶 液)各5ml,置25ml量瓶中,加甲醇稀释至 刻度,摇匀,取10μl注入液相色谱仪,记录
例
取盐酸麻黄碱0.1532g,精密称定, 加冰醋酸10ml溶解后,加醋酸汞试液 2ml与结晶紫指示液1滴,用HClO4滴 定至绿色,用去0.1022mol/L的高氯 酸滴定液7.50m1,空白试验消耗高氯 酸滴定液0.08ml。已知每1ml高氯酸 滴定液(0.1mol/L)当于20.17mg的 C10H15ON·HCl。试计算盐酸麻黄碱 的百分含量?(99.80%)
1.分离原理
吸附色谱 分配色谱 离子交换色谱 排阻色谱
吸附色谱
根据填充剂吸附活性 对样品的吸收系数不 同而分离
充填剂:硅胶
分配色谱
根据固定相与流动相的极性不同而分为 正相分配色谱和反相分配色谱 两者的区别如下图所示:
固定相
流动相
正相分配
极性
(2021年整理)8《药物分析》第四章(2课时)

武汉软件工程职业学院教案(理论教学首页)(第1页)章节名称第四章药物的杂质检查授课安排授课时数2授课时间第五周授课方法讲授授课教具多媒体教学目的1、明确药物的杂质“特殊物质的检查”的测定意义、原理及注意事项;2、学会测定药物中特殊杂质的操作技术。
教学重点学会测定药物中特殊杂质的操作技术;教学难点学会测定药物中特殊杂质原理及注意事项;项目6几种药物特殊杂质的检查一、利用药物与杂质在物理性质方面的差异(一)臭味及挥发性质的差异例如:黄凡士林中异性有机物检查:本品2.0g ,直火加热无辛味。
浓过氧化氢中不挥发性杂质检查:10ml 水浴蒸干残渣<15mg 。
(二)颜色差异例如:磺胺嘧啶中有色偶氮苯化合物的检查:取本品2.0g ,加NaOH 试液10ml ,加水25ml ,与黄色3号比色液比,不得更深。
(三)溶解行为的差异:例如:吡哌酸中碱中不溶物吡哌酸甲酯(I )和(II )(四)旋光性质的差异:例如:硫酸阿托品中莨菪碱检查:本品溶液(50mg/ml)α<-0.4(五)对光吸收性质的差异(1)紫外分光光度法:①在一定的波长杂质有吸收,而药物无吸收;②杂质杂质紫外吸收光谱与药物紫外吸收光谱重叠;③药物在紫外区有明显吸收,而杂质吸收很弱货物吸收。
(2)原子吸收分光光度法:主要用于药物中金属盐等杂质的检查,如曾用于维生素C 、硫酸庆大霉素和安痛定注射液中Na 、K 、Ca 、Mg 含量测定。
(3)红外分光光度法:主要用于药物中无效和低效晶型的检查,如甲苯米唑装订线中A晶型的检查。
(4)荧光分析法:例如利血平中氧化产物的检查,是根据利血平纯品无荧光,其氧化产物有荧光。
1.紫外分光光度法:例如:地蒽酚中二羟基蒽醌的检查。
再如:苯丙醇中苯丙酮的检查:可利用供试液的吸收度比来控制杂质;纯品苯丙醇A247nm/A258nm=0.59,99.5%时A247nm/A258nm=0.79;因此规定供试品A247nm/A258nm<0.79;即所含苯丙酮<0.5%。
第四章-药物定量分析和分析方法验证

方法分类
按分离方法分为:PC;TLC;柱色谱;GC;HPLC。
(一)HPLC法
1. 对HPLC仪器一般要求 色谱柱、流动相按品种项下要求。
2. 系统性试验
色谱柱的理论板数:n = 5.54(tR /Wh/2)2 分离度:R = 2(tR1 – tR2)/(W1 + W2); 要大于1.5 拖尾因子:T = W0.05h/2d1 应在0.9 ~ 1.05
后银量法
3. 利用药物中可游离的金属离子的氧化性测定含量
(1)含锑药物 例如:葡萄糖酸锑的含量测定
Sb5+ + 2KI H+ Sb3+ + 2K+ ++ I2
I2 + 2Na2S2O3
2NaI + Na2S4O6
(2)含铁药物
2Fe3+ + 2KI I2 + 2Na2S2O3
2Fe2+ + 2K+ + I2 2NaI + Na2S4O6
2. 制剂 要考察辅料对回收率的影响。采用在空白辅料中加入原 料对照品的方法作回收率试验,然后计算RSD。
具体做法:测定高、中、低三个浓度,n=3, 共9个数据来评价 回收率的RSD<2%;用UV和HPLC法时,一般回 收率可达98%~102%;容量法可达99.7%~100.3%
(二)数据要求
回收率% = (测定平均值— 空白值)/加入量×100% 要求制备高、中、低三浓度的样品,各测定3次。应 报告已知加入量的回收率,或测定结果平均值与真实值 之差及其可信限。
3. 百分含量的计算
(1)直接滴定法 D% = V ×F × T/W ×100% F = 实际标定的浓度/规定的浓度
药物分析-药物的杂质检查方法总结
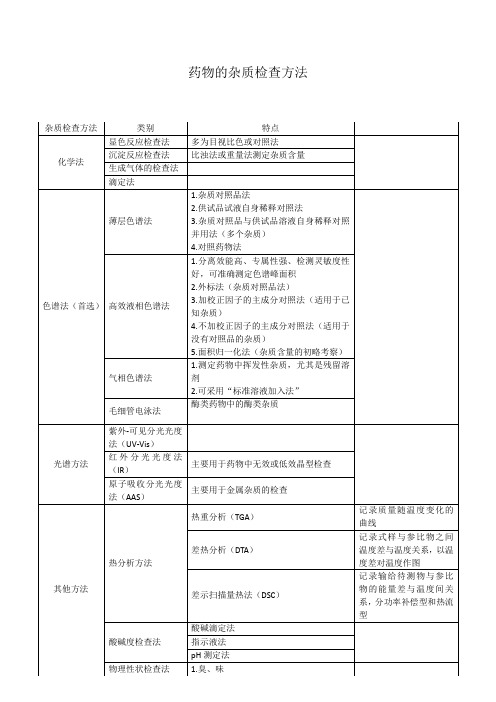
酸碱滴定法
指示液法
pH测定法
物理性状检查法
1.臭、味
2.挥发性
3.颜色
4.溶解性
5.其他物理属性(旋光性、黏度等)
红外分光光度法(IR)
主要用于药物中无效或低效晶型检查
原子吸收分光光度法(AAS)
主要用于金属杂质的检查
其他方法
热分析方法
热重分析(TGA)
记录质量随温度变化的曲线
差热分析(DTA)
记录式样与参比物之间温度差与温度关系,以温度差对温度作图
差示扫描量热法(DSC)
记录输给待测物与参比物的能量差与温度间关系,分功率补偿型和热流型
药物的杂质检查方法
杂质检查方法
类别
特点
化学法
显色反应检查法
多为目视比色或对照法
沉淀反应检查法
比浊法或重量法测定杂质含量
生成气体的检查法
滴定法
色谱法(首选)
薄层色谱法
1.杂质对照品法
2.供试品试液自身稀释对照法
3.杂质对照品与供试品溶液自身稀释对照并用法(多个杂质)
4.对照药物法
高效液相色谱法
1.分离效能高、专属性强、检测灵敏度性好,可准确测定色谱峰面积
2.外标法(杂质对照品法)
3.加校正因子的主成分对照法(适用于已知杂质)
4.不加校正因子的主成分对照法(适用于没有对照品的杂质)
5.面积归一化法(杂质含量的初略考察)
气相色谱法
1.测定药物中挥发性杂质,尤其是残留溶剂
2.可采用“标准溶液加入法”
毛细管电泳法
酶类药物中的Vis)
药物分析药物的检查

供
稀
释
试
对பைடு நூலகம்
品照
2. 灵敏度法
不需对照物质
系指在供试品溶液中加入试剂,在 一定反应条件下,不得有正反应出现
50ml H2O + 5滴HNO3 不浑浊
纯水中Cl-合格
3. 比较法
不需对照物质
含量测定法:测定杂质的绝对含 量,如测定吸收度、pH值等。
特点:准确测定杂质的量。
(三) 杂质限量的计算
杂质限量
例一. 药物中氯化物杂质检查,是使该 杂质在酸性溶液中与硝酸银作用生成 氯化物浑浊,所用的稀酸是(B)
A. 硫酸
B. 硝酸
C. 盐酸
D. 醋酸
E. 磷酸
例2. 中国药典 (2015年版) 规定,检 查氯化物杂质时,一般取用标准氯化 钠溶液(10µgCl-/ml)5~8ml的原因 是(D)
A. 使检查反应完全
三、杂质的分类
按来源分
一般杂质
特殊杂质
一般杂质:自然界是中分布较广,容易引入的物
质。其方法收载在中国药典的附录
中。 特殊杂质:指某一个或某一类药物的生产或贮藏
过程中引入的杂质。杂质检查方法收
载在中国药典正文各药品的质量标准
中。
例 阿司匹林 水杨酸
水解一步
COOH OCOCH3 + 2NaOH
COONa OH + CH3COONa
2.另取各药品项下规定量的标准氯 化钠溶液,置50ml纳氏比色管中,加 稀硝酸10m1,加水使成40m1,摇
匀,即得对照液。
3.于供试溶液与对照溶液中,分别 加入硝酸银试液1.0m1,用水稀释使 成50m1,摇匀,在暗处放置5分钟, 同置黑色背景上,从比色管上方向下 观察,比较,即得
药物分析课件 第四章 药物定量分析与分析方法验证_OK

③要有防爆措施 ④氧气要充足,确保燃烧完全,产物不能有黑色
炭化物 ⑤燃烧产生的烟雾要被完全吸收
充分振摇和放置都是为了保证吸收完全 例:碘产生紫色烟雾;氯、氟产生白色烟雾,烟
雾颜色完全消失后,即表示吸收完全 ⑥测定含氟有机化合物时要用石英燃烧瓶
26
4、应用
含卤素、硫、磷、氟及硒等有机药物的鉴别、杂 质的检查及含量测定
100%
注射剂:标示量%=
V0
V T 标示量
100%
V VO
T F 标示量
100%
VO : 量取注射剂的体积, ml
36
(2)间接滴定法 1)生成物滴定法
供试品+试剂 A 化合物 B
化合物 B+滴定液 C 化合物 D
计量滴定液消耗的量,计算供试品的含量 计算同直接滴定法
37
2)回滴定法(剩余滴定法) 原理:先在待测样品溶液中加定量、过量的滴定
10
(一)湿法破坏
(1) 硝酸-高氯酸法 适用于血、尿、组织等生物样品的破坏,有
机金属药物经破坏后,得到的无机金属离子一 般呈高价态。
本法不能用于含氮杂环药物的破坏
干法灼烧法 11
(2) 硝酸-硫酸法 适用于大多数有机物质的破坏,破坏得到的
无机金属离子均为高价态。 不能用于含碱土金属有机药物的破坏
27
氧瓶燃烧
第四章 药物的鉴别试验

第4章药物的鉴别试验依据药典进行的药物分析主要有三大项内容:鉴别、检查和含量测定。
药物的鉴别试验(ldentification test),是利用药物的分子结构所表现的特殊的化学行为(如进行化学反应、测定药物的理化常数等)或光谱、色谱、生物学等特征,来判断药品的真伪。
药物的鉴别在药品质量检验工作中属首项工作。
只有在药物被鉴别无误、证实被分析的药物是真的后,才有必要接着进行检查和含量测定等分析工作。
药典所收载的药物项下的鉴别试验方法,仅适用于贮藏在有标签容器中的药物,用以证实是否为其所标示的药物。
这些鉴别试验方法虽然具有一定的专属性,但是还不足以用来确证化合物的结构,因而一与分析化学中的定性鉴别有所区别,不能用来鉴别未知物。
因此,《中国药典》(2010年版)凡例中对药品“鉴别”项目的要求是:鉴别项下规定的试验方法,系根据反映该药品某些物理、化学或生物学等特征所进行的药物鉴别试验,不完全代表对该药品化学结构的确证。
本章讲授药物常用的方法和药物的一般鉴别试验,其中以化学鉴别法和仪器鉴别法为重点。
4.1鉴别试验的项目鉴别项下规定的实验方法,仅仅适用于鉴别药物的真伪。
对于原料药,还应该结合“性状”项下的外观和物理常数进行确认。
4.1.1性状(Description)药物的性状反映了药物特有的物理性质,一般包括外观、嗅、味、溶解度以及物理常数等。
性状观测是药品检验工作的第一步,也是不可省略的极其重要的一步。
只有性状符合规定的供试品,方可继续检查杂质限量和测定含量,否则不必进行检查和含量测定。
1.外观所谓药品的外观,是指药品的外表感官和色泽,包括药品的聚集状态、晶型、色泽以及臭、味等性质。
如《中国药典》(2010年版)对维生素A的描述为“本品为淡黄色溶液或结晶与油的混合物(加热至60℃应为澄清溶液);无臭;在空气中易氧化,遇光易变质。
”对于维生素AD的描述为“本品或本品内容物为黄色至橙红色的澄清油状液体;无败油臭或苦味”。
药物分析第四章定量分析与分析方法验证

标准偏差
三、专属性 系指在其它成份(如杂质、降解产物、辅料等) 系指在其它成份(如杂质、降解产物、辅料等)可能存在 采用的方法能准确测定出被物的特性。 下,采用的方法能准确测定出被物的特性。 四、检测限(LOD) 检测限( ) 指试样中被测物能被检出的最低浓度或量。 指试样中被测物能被检出的最低浓度或量。 五、定量限(LOQ) 定量限( ) 指样品中被测物能被定量测定的最低量, 指样品中被测物能被定量测定的最低量,其测定结果具有 一定的准确度和精密度。 一定的准确度和精密度。 六、线性 系指在设计的范围内, 系指在设计的范围内,测试结果与试样中被测物浓度直接 呈正比关系的程度,相关系数R-->1 ,用软件可得回归方程 用软件可得回归方程. 呈正比关系的程度,相关系数 用软件可得回归方程
VT 含量% = × 100% W
T—滴定度 滴定度;V—消耗滴定液体积 消耗滴定液体积;W—供试品取量 滴定度 消耗滴定液体积 供试品取量 校正因子F: 校正因子 当滴定液浓度与药典中规定浓度不相同时,要校正 要校正: 当滴定液浓度与药典中规定浓度不相同时 要校正 实际摩尔浓度 F= 规定摩尔浓度 在药检部门实验里的试剂瓶子一般不标浓度,而是 值 在药检部门实验里的试剂瓶子一般不标浓度 而是F值. 而是 实际滴定度:T’=TF 实际滴定度
二、光谱分析法 光照在物质上,物质内部发生能级跃迁而产生辐射, 光照在物质上,物质内部发生能级跃迁而产生辐射,利用 光谱进行定性或定量的结构分析称为光谱分析法。 光谱进行定性或定量的结构分析称为光谱分析法。 1、紫外-可见分光光度法(200nm—760nm) 、紫外 可见分光光度法 可见分光光度法( (1)朗伯 比耳定律 朗伯-比耳定律 朗伯 A=ELC A-吸光度;L-液层厚度;C-溶液浓度;E-吸收系数 吸光度; 液层厚度 液层厚度; 溶液浓度 溶液浓度; 吸收系数 吸光度 (2)百分吸收系数 )百分吸收系数E 1% 溶液浓度为1%( 溶液浓度为 (W/V)厚度为 )厚度为1cm的吸收度 E1cm 的吸收度 A (3)吸收系数法测含量 ) ×D 1% E1cm 含量%= ×100% W × l ×100 A-供试品吸光度;D-供试品稀释倍数; 供试品吸光度; 供试品稀释倍数 供试品稀释倍数; 供试品吸光度 W-供试品取量(标示量)( ); 液层厚度(cm) 供试品取量( )(g); 液层厚度( ) 供试品取量 标示量)( );L-液层厚度
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一、药物的纯度
1、定义:药物的纯净程度,又称药用 规格(最高杂质限量),因药品的纯度检 查主要针对杂质检查,因此杂质检查又 称纯度检查。
纯度检查
性 质
制剂检查
安全性
有效性
改变原料
需
改变工艺 检
新药研究 查
可能引入而
未规定的杂质
影 响 药 物 的 稳 定 性 和 疗 效
杂
无 毒 副 作 用
含量测定法:测定杂质的绝对
含量,如测定吸收度、pH值等。
特点:准确测定杂质的量。
(三) 杂质限量的计算
允许杂质存在的最大量 杂质限量 100% 供试品量
标准溶液体积 标准溶液浓度 杂质限量 供试品量
L=C标×V标/ms ×100%
检查某药物中的砷盐,取标准砷 溶液 2ml (每 1ml 相当于 1g 的 As )制 备标准砷斑,砷盐限量为0.00004%, 应取供试品的量为 A. 0.50g
B. 氯化物检查 C. 溶出度检查 D. 重金属检查 E. 砷盐检查
四、
药物的杂质限量与检查法
(一)杂质限量
指药物中允许杂质存在的最大 量,通常用百分之几或百万分之几
(ppm)来表示
杂质量 ≤ 杂质限量 < 杂质量
药品合格
药品不合格
(二)药物的杂质检查法
对照法
杂质 检查法
灵敏度
比较法
1. 对照法
B. 合成过程中产生的中间体或副产物分离不
净造成
C. 需加入的各种试剂产生吸附,共沉淀生成
混晶等造成
D. 所用金属器皿及装置等引入杂质
E. 由于操作不妥,日光曝晒而使产品发生分 解引入的杂质
三、杂质的分类
按来源分
一般杂质 特殊杂质
一般杂质:自然界是中分布较广,容易引入的物
质。其方法收载在中国药典的附录 中。 特殊杂质:指某一个或某一类药物的生产或贮藏 过程中引入的杂质。杂质检查方法收 载在中国药典正文各药品的质量标准 中。
适量稀释成50ml,摇匀;
如显色,立即与标准铁溶液一定量制
成的对照溶液(取各药品项下规定量 的标准铁溶液,臵50ml纳氏比色管中,
加水使成25ml,加稀盐酸4ml与过硫
酸铵50mg,用水稀释使成35m1后,加 30%硫氰酸铵溶液3ml,再加水适量稀
释成50ml,摇匀)比较,即得
3. 测定条件
(1)用FeNH4(SO4)2· 12H2O(硫酸铁
加稀硝酸10m1,加水使成40m1,摇匀,
即得对照液。
3.于供试溶液与对照溶液中,分 别加入硝酸银试液1.0m1,用水稀释使
成50m1,摇匀,在暗处放臵5分钟,同
臵黑色背景上,从比色管上方向下观 察,比较,即得
(三)测定条件
1. 标准NaCl溶液 10gCl/ml,50ml
溶液中含50~80g的Cl所显浑浊梯度 明显,相当于标准NaCl溶液5~8ml。
对照:Fe c、V 6SCN FeSCN 6 红色
3 HCl 3
2. 检查方法 药典附录
除另有规定外,取各药品项下规 定量的供试品,加水溶解使成25ml,
移臵50ml纳氏比色管中,加稀盐酸4ml
与过硫酸铵50mg,用水稀释使成 35m1 后,加30%硫氰酸铵溶液3ml,再加水
D. 用硝酸银标准溶液做对照
E. 在白色背景下观察
三、硫酸盐检查法
(一)原理 对照法
药物:SO2 4
BaCl2 BaSO4白色浑浊
HCl
HCl
对照:K 2SO( BaCl2 BaSO4白色浑浊 4 c、V)
(二)测定条件
1. 标准K2SO4溶液0.1mg/ml,50ml溶
2. 反应需在硝酸酸性条件下进行, 且以50ml供试溶液中含稀硝酸10ml为
宜。
(1)加速AgCl浑浊的形成;
(2)产生较好的乳浊; (3)避免弱酸银盐如碳酸银、磷酸银 以及氧化银沉淀的形成。
3. 试剂:硝酸银 4. 供试液和对照液稀释后,再加硝酸 银溶液,使生成白色浑浊而不是白 色沉淀 5. 避光、暗处放臵5分钟后比浊,因氯 化银见光易分解。 6. 比浊方法:同臵于黑色背景上,自 上向下观察。 7. 平行操作原则
A. 使检查反应完全 B. 药物中含氯化物的量均在此范围
C. 加速反应
D. 所产生的浊度梯度明显
E. 避免干扰
例3. 采用硝酸银试液检查氯化物时, 加入硝酸使溶液酸化的目的是 (ABCDE)
A.加速生成氯化银浑浊反应
B.消除某些弱酸盐的干扰
C.消除碳酸盐干扰 D.消除磷酸盐干扰
E.避免氧化银沉淀生成
氰酸铵,使反应向正反应方向进行 (4)检查Fe2+和Fe3+
(5)铁盐检查时,加氧化剂氧化 Fe2+ 为 Fe3+ 。加过硫酸铵可氧化 Fe2+ 为
Fe3+,同时可防止光线使硫氰酸铁还原
或分解褪色。 加硝酸后加热也可氧化Fe2+为Fe3+
( 6 )比色方法:同臵于白色背景 上,自上向下观察。
4. 干扰及排除
液中含0.1~0.5mg的所显浑浊梯度明显,
相当于标准K2SO4溶液1~5ml。
2.反应需在盐酸酸性条件下进行,且 以50ml供试溶液中含稀盐酸2ml为宜。
3. 试剂:氯化钡
4. 供试液和对照液稀释后,再加
氯化钡溶液,使生成白色浑浊而不是 白色沉淀
5. 比浊方法:同臵于黑色背景上
自上向下观察。
(三)干扰及排除
巯基醋酸
红色
对照: Fe
NH 3 H 2 O 3
Fe
巯基醋酸
20%枸橼酸 2 ml
巯基醋酸
2
红色
本法灵敏度较高,但试剂较贵
例1. 中国药典(2015年版)规定铁盐 的检查方法为(A) A. 硫氰酸盐法
B. 巯基醋酸法 C. 普鲁士蓝法
临用前稀释而成,标准硝酸铅标准溶液
10g Pb2+/ml,适宜比色范围为27ml溶 液中含10~20g的Pb2+。
(2)本法用2ml pH3.5的醋酸盐
缓冲液控制溶液pH值为3~3.5。 (3)显色剂:从ChP(1990)开 始改用硫代乙酰胺做显色剂
3. 干扰及排除
(1)供试品如有色,需经处理后方可检查 外消色法:在对照管中加稀焦糖溶液或
2. 正确的取样及供试品的称量范围
1g不超过±2%,>1g不超过±1% 3. 正确的比色、比浊方法 4. 检查结果不符合规定或在限度边缘
时应对供试管和对照管各复查二份
二、氯化物检查法
(一)原理 对照法
药物:Cl AgNO3 AgCl白色浑浊
对照:NaCl(c,V) AgNO3 AgCl白色浑浊
(1)为了提高灵敏度或供试管与对照
管色调不一致时,可用正丁醇提取后,
分取正丁醇层比色。
(2)具环状结构的有机药物,需经
700~800℃炽灼破坏处理后再依法检查。
(二)巯基醋酸法
Fe 3 药物: Fe 2 NH 3 H 2O 20%枸橼酸 2 ml
对照法
巯基醋酸 2
Fe
HNO 3
HNO 3
(二)检查方法 药典附录
1.除另有规定外,取各药品项下 规定量的供试品,加水溶解使成25ml
(溶液如显碱性,可滴加硝酸使成中
性),再加稀硝酸10m1;溶液如不澄
清,应滤过;臵50ml纳氏比色管中,
加水使成约40m1,摇匀,即得供试溶 液。
2.另取各药品项下规定量的标准 氯化钠溶液,臵50ml纳氏比色管中,
若供试品有色,也可用内消色法
即倍量法处理。
例:药物中硫酸盐检查时,所用的标
准对照液是(D)
A. 标准氯化钡
B. 标准醋酸铅溶液 C. 标准硝酸银溶液
D. 标准硫酸钾溶液 E. 以上都不对
四、铁盐检查法
(一)硫氰酸盐法 ChP(2005)、USP(24) 1. 原理 对照法
o 3 Fe 2 HCl Fe 3 红色 药物: 6SCN Fe SCN 3 6 Fe
(四)干扰及排除
(1)内消色法:倍量法,如枸橼酸铁铵
1. 若供试品有色,需经处理后方可检查。
中氯化物的检查。在对照液中加一定量供
试液。
(2)外消色法:如高锰酸钾中氯化物的
检查,可先加乙醇适量,使其还原褪色后
再依法检查。
2. 不溶于水的有机药物
(1)加水振摇,过滤,取滤液进行 检查。
(2)加热,放冷,过滤,取滤液进
pH3.5
药物: Pb H2S PbS黄色~棕黑色
pH3.5
pH3.5 对照:Pb(NO ) 2 c 、 V H S PbS黄色~棕黑色 3 2
2
适用于溶于水、稀酸和乙醇的药物。
2. 测定条件
(1)用硝酸铅配制标准铅贮备液 (加硝酸防止Pb2+水解),标准铅溶液
A. Fe
B. Fe2+
C. Fe3+ D. Fe2+和Fe3+
E. 以上都不对
五、重金属检查法
第一法 硫代乙酰胺法
以铅为代表
中国药典(2015年版)共收载四法。 第二法 第三法
第四法
炽灼残渣法 硫化钠法
微孔滤膜法
(一)第一法 硫代乙酰胺法 1. 原理 对照法
CH3CSNH 2 H2O CH3CONH2 H2S
例4. 当采用比浊法检查氯化物杂质时,
若药物本身有颜色而干扰检查的话,
应该选用的处理方法为(AB)
A.内消色法 B.外消色法 C.比色法 D.差示比浊法