Hyperchem学习
化学信息学——第八讲hyperchem 软件学习

HyperChem程序简介 7.5版新增功能 OpenGL绘图: 基础演示模式:已全部转换为全新的 OpenGL模式,使分子演示图形具有更高 质量支持自定义色彩,1600万种颜色代替 了传统的8种标准色。 混合演示:棒型,球型或组合型等分子 演示手段可以用于该分子的任意原子及链 段的演示,及全新的适用于原子的"管型" 演示。增强的蛋白质模型构建功能,支持 多种蛋白质二级结构演示,大分子的电子 密度近似方法。
13
工具 条 •使用图标工具可以简化操作 •从左至右分别是绘制、选择、旋转、平面 转动、平移
14
工作状态
•工作 区: •状态 行:
15
2、打开已存在的数据文件 •File-Open
16
浏览文件夹
17
C60分子
18
选择分子图形的显示方式 •Display- Labels可以选择原子、化学键等 标记方式:
39
计算几何构型 •COMPUTE---GEOMETRY
40
计算结果 •优化结束
41
测量键长、键角、二面角等结构参数 •数值显示在底部状态栏
42
单点能计算
•对给定体系优化构型后,可对优化后的构 型再进行单点计算。
43
单点能结果 •单点能结果
44
输出结果
45
计算结果的输出文件
• • • • • • • • • • • • • • • HyperChemlog start -- Sun May 28 21:38:15 2000. Geometry optimization, SemiEmpirical, molecule = (untitled). CNDO PolakRibiere optimizer Convergence limit = 0.0001000 Iteration limit = 50 Accelerate convergence = YES Optimization algorithm = Polak-Ribiere Criterion of RMS gradient = 0.0100 kcal/(A mol) Maximum cycles = 150 RHF Calculation: Singlet state calculation Number of electrons = 22 Number of Double Occupied Levels = 11 Charge on the System = 0 Total Orbitals = 22
hyperchem应用-辛烷

Hyperchem程序应用-辛烷1、画辛烷分子模型:辛烷分子碳碳骨架结构:加氢并模型化:2、用半经验方法CNDO进行优化3、选用从头算计算方法进行单点计算:4、显示原子电荷、显示键长、显示键角、二面角:5、分子性质:6、分子轨道图-最高占据轨道2D、3D图:7、分子轨道图-最低空轨道2D、3D图:8辛烷分子总电荷密度图(2D、3D):8、辛烷分子2D、3D静电势图、等值面图:9、分子结构模型表示:10、计算输出结果:HyperChem log start -- Mon Dec 13 18:37:31 2010.Geometry optimization, SemiEmpirical, molecule = (untitled).CNDOFletcherReeves optimizerConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESOptimization algorithm = Fletcher-ReevesCriterion of RMS gradient = 0.1000 kcal/(A mol) Maximum cycles = 345RHF Calculation:Singlet state calculationNumber of electrons = 44Number of Double Occupied Levels = 22Charge on the System = 0Total Orbitals = 44Starting CNDO calculation with 44 orbitalsE=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=14292.75153]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=2 Diff=9.35887]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=3 Diff=0.54071]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=4 Diff=0.03993]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=5 Diff=0.00066]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=6 Diff=0.00004]E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=1 Diff=115.68581] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=2 Diff=9.46473] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=3 Diff=0.86416] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=4 Diff=0.10244] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=5 Diff=0.00180] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=6 Diff=0.00012] E=-5569.6934 Grad=74.633 Conv=NO(0 cycles 1 points) [Iter=7 Diff=0.00001] E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=1 Diff=46.41182] E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=2 Diff=3.63947]E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=3 Diff=0.32064] E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=4 Diff=0.03685] E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=5 Diff=0.00059] E=-5516.4321 Grad=113.901 Conv=NO(0 cycles 2 points) [Iter=6 Diff=0.00003] E=-5613.0083 Grad=6.496 Conv=NO(1 cycles 3 points) [Iter=1 Diff=0.06673] E=-5613.0083 Grad=6.496 Conv=NO(1 cycles 3 points) [Iter=2 Diff=0.00512] E=-5613.0083 Grad=6.496 Conv=NO(1 cycles 3 points) [Iter=3 Diff=0.00050] E=-5613.0083 Grad=6.496 Conv=NO(1 cycles 3 points) [Iter=4 Diff=0.00007] E=-5613.5781 Grad=3.499 Conv=NO(1 cycles 4 points) [Iter=1 Diff=0.06718] E=-5613.5781 Grad=3.499 Conv=NO(1 cycles 4 points) [Iter=2 Diff=0.00516] E=-5613.5781 Grad=3.499 Conv=NO(1 cycles 4 points) [Iter=3 Diff=0.00050] E=-5613.5781 Grad=3.499 Conv=NO(1 cycles 4 points) [Iter=4 Diff=0.00007] E=-5613.6587 Grad=4.830 Conv=NO(1 cycles 5 points) [Iter=1 Diff=0.00755] E=-5613.6587 Grad=4.830 Conv=NO(1 cycles 5 points) [Iter=2 Diff=0.00058] E=-5613.6587 Grad=4.830 Conv=NO(1 cycles 5 points) [Iter=3 Diff=0.00006] E=-5613.6865 Grad=3.874 Conv=NO(2 cycles 6 points) [Iter=1 Diff=0.09278] E=-5613.6865 Grad=3.874 Conv=NO(2 cycles 6 points) [Iter=2 Diff=0.00694] E=-5613.6865 Grad=3.874 Conv=NO(2 cycles 6 points) [Iter=3 Diff=0.00063] E=-5613.6865 Grad=3.874 Conv=NO(2 cycles 6 points) [Iter=4 Diff=0.00008] E=-5613.8223 Grad=3.477 Conv=NO(2 cycles 7 points) [Iter=1 Diff=0.00902] E=-5613.8223 Grad=3.477 Conv=NO(2 cycles 7 points) [Iter=2 Diff=0.00067] E=-5613.8223 Grad=3.477 Conv=NO(2 cycles 7 points) [Iter=3 Diff=0.00006] E=-5613.8574 Grad=2.075 Conv=NO(3 cycles 8 points) [Iter=1 Diff=0.00837] E=-5613.8574 Grad=2.075 Conv=NO(3 cycles 8 points) [Iter=2 Diff=0.00083] E=-5613.8574 Grad=2.075 Conv=NO(3 cycles 8 points) [Iter=3 Diff=0.00010] E=-5613.8574 Grad=2.075 Conv=NO(3 cycles 8 points) [Iter=4 Diff=0.00002] E=-5613.9468 Grad=1.293 Conv=NO(3 cycles 9 points) [Iter=1 Diff=0.00837] E=-5613.9468 Grad=1.293 Conv=NO(3 cycles 9 points) [Iter=2 Diff=0.00083] E=-5613.9468 Grad=1.293 Conv=NO(3 cycles 9 points) [Iter=3 Diff=0.00010] E=-5613.9468 Grad=1.293 Conv=NO(3 cycles 9 points) [Iter=4 Diff=0.00002] E=-5613.9312 Grad=2.539 Conv=NO(3 cycles 10 points) [Iter=1 Diff=0.00351] E=-5613.9312 Grad=2.539 Conv=NO(3 cycles 10 points) [Iter=2 Diff=0.00035] E=-5613.9312 Grad=2.539 Conv=NO(3 cycles 10 points) [Iter=3 Diff=0.00004] E=-5613.9531 Grad=1.581 Conv=NO(4 cycles 11 points) [Iter=1 Diff=0.03067] E=-5613.9531 Grad=1.581 Conv=NO(4 cycles 11 points) [Iter=2 Diff=0.00315] E=-5613.9531 Grad=1.581 Conv=NO(4 cycles 11 points) [Iter=3 Diff=0.00041] E=-5613.9531 Grad=1.581 Conv=NO(4 cycles 11 points) [Iter=4 Diff=0.00007] E=-5614.0254 Grad=1.729 Conv=NO(4 cycles 12 points) [Iter=1 Diff=0.03082] E=-5614.0254 Grad=1.729 Conv=NO(4 cycles 12 points) [Iter=2 Diff=0.00317] E=-5614.0254 Grad=1.729 Conv=NO(4 cycles 12 points) [Iter=3 Diff=0.00041] E=-5614.0254 Grad=1.729 Conv=NO(4 cycles 12 points) [Iter=4 Diff=0.00007] E=-5613.9551 Grad=3.772 Conv=NO(4 cycles 13 points) [Iter=1 Diff=0.03047] E=-5613.9551 Grad=3.772 Conv=NO(4 cycles 13 points) [Iter=2 Diff=0.00314] E=-5613.9551 Grad=3.772 Conv=NO(4 cycles 13 points) [Iter=3 Diff=0.00041]E=-5614.0254 Grad=1.739 Conv=NO(5 cycles 14 points) [Iter=1 Diff=0.10421] E=-5614.0254 Grad=1.739 Conv=NO(5 cycles 14 points) [Iter=2 Diff=0.00961] E=-5614.0254 Grad=1.739 Conv=NO(5 cycles 14 points) [Iter=3 Diff=0.00112] E=-5614.0254 Grad=1.739 Conv=NO(5 cycles 14 points) [Iter=4 Diff=0.00018] E=-5614.0254 Grad=1.739 Conv=NO(5 cycles 14 points) [Iter=5 Diff=0.00000] E=-5614.0059 Grad=2.873 Conv=NO(5 cycles 15 points) [Iter=1 Diff=0.02966] E=-5614.0059 Grad=2.873 Conv=NO(5 cycles 15 points) [Iter=2 Diff=0.00274] E=-5614.0059 Grad=2.873 Conv=NO(5 cycles 15 points) [Iter=3 Diff=0.00032] E=-5614.0059 Grad=2.873 Conv=NO(5 cycles 15 points) [Iter=4 Diff=0.00005] E=-5614.0864 Grad=0.979 Conv=NO(6 cycles 16 points) [Iter=1 Diff=0.00695] E=-5614.0864 Grad=0.979 Conv=NO(6 cycles 16 points) [Iter=2 Diff=0.00061] E=-5614.0864 Grad=0.979 Conv=NO(6 cycles 16 points) [Iter=3 Diff=0.00007] E=-5614.1025 Grad=0.870 Conv=NO(6 cycles 17 points) [Iter=1 Diff=0.00052] E=-5614.1025 Grad=0.870 Conv=NO(6 cycles 17 points) [Iter=2 Diff=0.00005] E=-5614.1055 Grad=0.577 Conv=NO(7 cycles 18 points) [Iter=1 Diff=0.00479] E=-5614.1055 Grad=0.577 Conv=NO(7 cycles 18 points) [Iter=2 Diff=0.00048] E=-5614.1055 Grad=0.577 Conv=NO(7 cycles 18 points) [Iter=3 Diff=0.00006] E=-5614.1069 Grad=0.931 Conv=NO(7 cycles 19 points) [Iter=1 Diff=0.00096] E=-5614.1069 Grad=0.931 Conv=NO(7 cycles 19 points) [Iter=2 Diff=0.00010] E=-5614.1108 Grad=0.447 Conv=NO(8 cycles 20 points) [Iter=1 Diff=0.00213] E=-5614.1108 Grad=0.447 Conv=NO(8 cycles 20 points) [Iter=2 Diff=0.00018] E=-5614.1108 Grad=0.447 Conv=NO(8 cycles 20 points) [Iter=3 Diff=0.00002] E=-5614.1177 Grad=0.476 Conv=NO(8 cycles 21 points) [Iter=1 Diff=0.00213] E=-5614.1177 Grad=0.476 Conv=NO(8 cycles 21 points) [Iter=2 Diff=0.00018] E=-5614.1177 Grad=0.476 Conv=NO(8 cycles 21 points) [Iter=3 Diff=0.00002] E=-5614.1167 Grad=0.939 Conv=NO(8 cycles 22 points) [Iter=1 Diff=0.00085] E=-5614.1167 Grad=0.939 Conv=NO(8 cycles 22 points) [Iter=2 Diff=0.00007] E=-5614.1182 Grad=0.627 Conv=NO(9 cycles 23 points) [Iter=1 Diff=0.02140] E=-5614.1182 Grad=0.627 Conv=NO(9 cycles 23 points) [Iter=2 Diff=0.00201] E=-5614.1182 Grad=0.627 Conv=NO(9 cycles 23 points) [Iter=3 Diff=0.00022] E=-5614.1182 Grad=0.627 Conv=NO(9 cycles 23 points) [Iter=4 Diff=0.00003] E=-5614.1118 Grad=1.125 Conv=NO(9 cycles 24 points) [Iter=1 Diff=0.00758] E=-5614.1118 Grad=1.125 Conv=NO(9 cycles 24 points) [Iter=2 Diff=0.00071] E=-5614.1118 Grad=1.125 Conv=NO(9 cycles 24 points) [Iter=3 Diff=0.00008] E=-5614.1235 Grad=0.266 Conv=NO(10 cycles 25 points) [Iter=1 Diff=0.00049] E=-5614.1235 Grad=0.266 Conv=NO(10 cycles 25 points) [Iter=2 Diff=0.00005] E=-5614.1260 Grad=0.299 Conv=NO(10 cycles 26 points) [Iter=1 Diff=0.00049] E=-5614.1260 Grad=0.299 Conv=NO(10 cycles 26 points) [Iter=2 Diff=0.00005] E=-5614.1245 Grad=0.624 Conv=NO(10 cycles 27 points) [Iter=1 Diff=0.00036] E=-5614.1245 Grad=0.624 Conv=NO(10 cycles 27 points) [Iter=2 Diff=0.00004] E=-5614.1260 Grad=0.339 Conv=NO(11 cycles 28 points) [Iter=1 Diff=0.00379] E=-5614.1260 Grad=0.339 Conv=NO(11 cycles 28 points) [Iter=2 Diff=0.00040] E=-5614.1260 Grad=0.339 Conv=NO(11 cycles 28 points) [Iter=3 Diff=0.00005]E=-5614.1328 Grad=0.318 Conv=NO(11 cycles 29 points) [Iter=2 Diff=0.00040] E=-5614.1328 Grad=0.318 Conv=NO(11 cycles 29 points) [Iter=3 Diff=0.00005] E=-5614.1348 Grad=0.524 Conv=NO(11 cycles 30 points) [Iter=1 Diff=0.01513] E=-5614.1348 Grad=0.524 Conv=NO(11 cycles 30 points) [Iter=2 Diff=0.00160] E=-5614.1348 Grad=0.524 Conv=NO(11 cycles 30 points) [Iter=3 Diff=0.00020] E=-5614.1348 Grad=0.524 Conv=NO(11 cycles 30 points) [Iter=4 Diff=0.00003] E=-5614.1270 Grad=1.092 Conv=NO(11 cycles 31 points) [Iter=1 Diff=0.01436] E=-5614.1270 Grad=1.092 Conv=NO(11 cycles 31 points) [Iter=2 Diff=0.00152] E=-5614.1270 Grad=1.092 Conv=NO(11 cycles 31 points) [Iter=3 Diff=0.00019] E=-5614.1270 Grad=1.092 Conv=NO(11 cycles 31 points) [Iter=4 Diff=0.00003] E=-5614.1348 Grad=0.538 Conv=NO(12 cycles 32 points) [Iter=1 Diff=0.14614] E=-5614.1348 Grad=0.538 Conv=NO(12 cycles 32 points) [Iter=2 Diff=0.01365] E=-5614.1348 Grad=0.538 Conv=NO(12 cycles 32 points) [Iter=3 Diff=0.00158] E=-5614.1348 Grad=0.538 Conv=NO(12 cycles 32 points) [Iter=4 Diff=0.00025] E=-5614.1348 Grad=0.538 Conv=NO(12 cycles 32 points) [Iter=5 Diff=0.00000] E=-5613.9834 Grad=4.088 Conv=NO(12 cycles 33 points) [Iter=1 Diff=0.11050] E=-5613.9834 Grad=4.088 Conv=NO(12 cycles 33 points) [Iter=2 Diff=0.01032] E=-5613.9834 Grad=4.088 Conv=NO(12 cycles 33 points) [Iter=3 Diff=0.00120] E=-5613.9834 Grad=4.088 Conv=NO(12 cycles 33 points) [Iter=4 Diff=0.00019] E=-5613.9834 Grad=4.088 Conv=NO(12 cycles 33 points) [Iter=5 Diff=0.00000] E=-5614.1387 Grad=0.255 Conv=NO(13 cycles 34 points) [Iter=1 Diff=0.00190] E=-5614.1387 Grad=0.255 Conv=NO(13 cycles 34 points) [Iter=2 Diff=0.00021] E=-5614.1387 Grad=0.255 Conv=NO(13 cycles 34 points) [Iter=3 Diff=0.00003] E=-5614.1421 Grad=0.289 Conv=NO(13 cycles 35 points) [Iter=1 Diff=0.00190] E=-5614.1421 Grad=0.289 Conv=NO(13 cycles 35 points) [Iter=2 Diff=0.00021] E=-5614.1421 Grad=0.289 Conv=NO(13 cycles 35 points) [Iter=3 Diff=0.00003] E=-5614.1431 Grad=0.532 Conv=NO(13 cycles 36 points) [Iter=1 Diff=0.00010] E=-5614.1431 Grad=0.532 Conv=NO(13 cycles 36 points) [Iter=2 Diff=0.00001] E=-5614.1431 Grad=0.470 Conv=NO(14 cycles 37 points) [Iter=1 Diff=0.03041] E=-5614.1431 Grad=0.470 Conv=NO(14 cycles 37 points) [Iter=2 Diff=0.00300] E=-5614.1431 Grad=0.470 Conv=NO(14 cycles 37 points) [Iter=3 Diff=0.00038] E=-5614.1431 Grad=0.470 Conv=NO(14 cycles 37 points) [Iter=4 Diff=0.00007] E=-5614.1255 Grad=1.489 Conv=NO(14 cycles 38 points) [Iter=1 Diff=0.01674] E=-5614.1255 Grad=1.489 Conv=NO(14 cycles 38 points) [Iter=2 Diff=0.00165] E=-5614.1255 Grad=1.489 Conv=NO(14 cycles 38 points) [Iter=3 Diff=0.00021] E=-5614.1255 Grad=1.489 Conv=NO(14 cycles 38 points) [Iter=4 Diff=0.00004] E=-5614.1455 Grad=0.166 Conv=NO(15 cycles 39 points) [Iter=1 Diff=0.00113] E=-5614.1455 Grad=0.166 Conv=NO(15 cycles 39 points) [Iter=2 Diff=0.00010] E=-5614.1455 Grad=0.166 Conv=NO(15 cycles 39 points) [Iter=3 Diff=0.00001] E=-5614.1465 Grad=0.312 Conv=NO(15 cycles 40 points) [Iter=1 Diff=0.00011] E=-5614.1465 Grad=0.312 Conv=NO(15 cycles 40 points) [Iter=2 Diff=0.00001] E=-5614.1465 Grad=0.207 Conv=NO(16 cycles 41 points) [Iter=1 Diff=0.00191] E=-5614.1465 Grad=0.207 Conv=NO(16 cycles 41 points) [Iter=2 Diff=0.00016]E=-5614.1465 Grad=0.338 Conv=NO(16 cycles 42 points) [Iter=1 Diff=0.00053]E=-5614.1465 Grad=0.338 Conv=NO(16 cycles 42 points) [Iter=2 Diff=0.00005]E=-5614.1470 Grad=0.118 Conv=NO(17 cycles 43 points) [Iter=1 Diff=0.00009]E=-5614.1475 Grad=0.087 Conv=NO(17 cycles 44 points) [Iter=1 Diff=0.00010]E=-5614.1479 Grad=0.135 Conv=NO(17 cycles 45 points) [Iter=1 Diff=0.00000]E=-5614.1479 Grad=0.125 Conv=NO(18 cycles 46 points) [Iter=1 Diff=0.00045]E=-5614.1479 Grad=0.125 Conv=NO(18 cycles 46 points) [Iter=2 Diff=0.00004]E=-5614.1475 Grad=0.250 Conv=NO(18 cycles 47 points) [Iter=1 Diff=0.00015]E=-5614.1475 Grad=0.250 Conv=NO(18 cycles 47 points) [Iter=2 Diff=0.00001]E=-5614.1479 Grad=0.080 Conv=YES(19 cycles 48 points) [Iter=1 Diff=0.00000] ENERGIES AND GRADIENTTotal Energy = -39107.4530564 (kcal/mol)Total Energy = -62.5 (a.u.)Binding Energy = -5614.1480863 (kcal/mol)Isolated Atomic Energy = -33493.3049701 (kcal/mol)Electronic Energy = -151855.7014212 (kcal/mol)Core-Core Interaction = 112748.2483647 (kcal/mol)Heat of Formation = -3584.2860863 (kcal/mol)Gradient = 0.0797574 (kcal/mol/Ang) MOLECULAR POINT GROUPC2VEIGENV ALUES(eV)Symmetry: 1 A1 1 B2 2 A1 2 B2 3 A1 Eigenvalue: -48.228653 -44.921967 -39.969822 -34.146118 -30.883162 Symmetry: 1 B1 4 A1 3 B2 1 A2 5 A1 Eigenvalue: -28.682699 -28.469046 -28.056545 -25.610565 -24.098553 Symmetry: 4 B2 2 B1 5 B2 6 A1 2 A2 Eigenvalue: -23.099024 -21.719042 -20.119864 -18.474934 -18.154465 Symmetry: 7 A1 3 B1 6 B2 3 A2 8 A1 Eigenvalue: -16.890295 -15.730311 -15.627211 -14.505156 -14.398620 Symmetry: 7 B2 4 B1 5 B1 4 A2 9 A1 Eigenvalue: -14.220551 -14.122036 6.037422 6.486364 6.984152 Symmetry: 6 B1 8 B2 10 A1 9 B2 11 A1 Eigenvalue: 7.209129 7.266216 7.494792 7.807704 7.893167 Symmetry: 10 B2 5 A2 12 A1 11 B2 13 A1 Eigenvalue: 8.082231 8.194494 8.242883 8.928360 9.149508 Symmetry: 7 B1 14 A1 6 A2 12 B2 8 B1 Eigenvalue: 9.358259 9.783697 10.395283 10.967291 11.024303 Symmetry: 13 B2 15 A1 16 A1 14 B2Eigenvalue: 11.812114 11.856253 12.302794 12.332813ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C1.017689 0.970367 1.030565 0.973715 0.968621AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C1.012650 1.023395 0.956421 0.974890 1.013266AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C1.016258 0.960073 0.976386 1.014955 1.017676AO: 4 Pz C 5 S C 5 Px C 5 Py C 5 Pz C0.959603 0.974890 1.014978 1.014546 0.960073AO: 6 S C 6 Px C 6 Py C 6 Pz C 7 S C0.968622 1.017895 1.018150 0.956422 1.017688AO: 7 Px C 7 Py C 7 Pz C 8 S H 9 S H1.024900 0.976031 0.973714 1.011286 1.004890AO: 10 S H 11 S H 12 S H 13 S H 14 S H1.004890 1.014996 1.014996 1.015570 1.015570AO: 15 S H 16 S H 17 S H 18 S H 19 S H1.015582 1.015582 1.015570 1.015570 1.014996AO: 20 S H 21 S H 22 S H 23 S H1.014995 1.011287 1.004890 1.004890NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 0.007665 1.27026 -2.99875 0.00000 12.011002 6 0.038912 1.22452 -1.53312 0.00000 12.011003 6 0.035512 -0.13502 -0.95607 0.00000 12.011004 6 0.031379 -0.21279 0.51953 0.00000 12.011005 6 0.035513 -1.57806 1.08470 -0.00000 12.011006 6 0.038911 -1.66895 2.55885 0.00000 12.011007 6 0.007667 -3.03551 3.09050 -0.00000 12.011008 1 -0.011286 2.31779 -3.39733 -0.00000 1.008009 1 -0.004890 0.76635 -3.44042 0.89791 1.0080010 1 -0.004890 0.76635 -3.44042 -0.89790 1.0080011 1 -0.014996 1.80141 -1.14693 0.88746 1.0080012 1 -0.014996 1.80141 -1.14693 -0.88746 1.0080013 1 -0.015570 -0.70523 -1.35564 0.88637 1.0080014 1 -0.015570 -0.70523 -1.35564 -0.88637 1.0080015 1 -0.015582 0.35562 0.92145 0.88644 1.0080016 1 -0.015582 0.35562 0.92145 -0.88643 1.0080017 1 -0.015570 -2.14485 0.68030 -0.88638 1.0080018 1 -0.015570 -2.14486 0.68029 0.88636 1.0080019 1 -0.014996 -1.11255 2.97401 0.88746 1.0080020 1 -0.014995 -1.11254 2.97402 -0.88745 1.0080021 1 -0.011287 -3.06212 4.21099 0.00000 1.0080022 1 -0.004890 -3.61989 2.76263 -0.89791 1.0080023 1 -0.004890 -3.61990 2.76263 0.89790 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 -0.23321 0.17204 -0.000102 6 -0.05131 -0.05641 -0.000493 6 -0.12340 0.09713 -0.000494 6 0.05576 0.06527 -0.000275 6 0.07533 -0.18029 0.000046 6 -0.08613 -0.00112 -0.000097 6 0.10150 -0.28460 -0.000148 1 0.04407 0.00197 -0.000029 1 -0.01228 0.07302 0.0311010 1 -0.01224 0.07324 -0.0309911 1 -0.05895 -0.04436 -0.1303212 1 -0.05946 -0.04468 0.1308613 1 0.06288 0.03734 -0.0218714 1 0.06334 0.03765 0.0223715 1 0.05153 0.03609 0.1191916 1 0.05140 0.03583 -0.1189617 1 0.05580 0.04741 0.0218218 1 0.05573 0.04738 -0.0218019 1 -0.06188 -0.04176 -0.1312320 1 -0.06184 -0.04172 0.1312321 1 0.01654 0.04341 -0.0000122 1 0.06347 -0.03639 -0.0327523 1 0.06334 -0.03647 0.03294Dipole (Debyes) x y z TotalPoint-Chg. -0.024 -0.017 -0.000 0.030sp Hybrid 0.011 0.008 -0.000 0.013pd Hybrid 0.000 0.000 0.000 0.000Sum -0.013 -0.009 -0.000 0.016Single Point, AbInitio, molecule = (untitled).Convergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESFull MP2 correlation energy is requested.The initial guess of the MO coefficients is from eigenvectors of the core Hamiltonian. Shell Types: S, S=P.RHF Calculation:Singlet state calculationNumber of electrons = 58Number of Doubly-Occupied Levels = 29Charge on the System = 0Total Orbitals (Basis Functions) = 95Primitive Gaussians = 153Starting HyperGauss calculation with 95 basis functions and 153 primitive Gaussians. 2-electron Integral buffers will be 3200 words (double precision) long.Two electron integrals will use a cutoff of 1.00000e-010Regular integral format is used.Computing the one-electron integrals ...Computing 2e integrals (s and p orbitals only): done 0%.Computing 2e integrals (s and p orbitals only): done 10%.Computing 2e integrals (s and p orbitals only): done 20%.Computing 2e integrals (s and p orbitals only): done 30%.Computing 2e integrals (s and p orbitals only): done 40%.Computing 2e integrals (s and p orbitals only): done 50%.Computing 2e integrals (s and p orbitals only): done 60%.Computing 2e integrals (s and p orbitals only): done 70%.Computing 2e integrals (s and p orbitals only): done 80%.Computing 2e integrals (s and p orbitals only): done 90%.5631113 integrals have been produced.Computing the initial guess of the MO coefficients ...Iteration = 1 Difference = 1032.07Iteration = 2 Difference = 1938.56Iteration = 3 Difference = 189.33Iteration = 4 Difference = 632.45Iteration = 5 Difference = 171.79Iteration = 6 Difference = 24.01Iteration = 7 Difference = 2.02Iteration = 8 Difference = 0.35Iteration = 9 Difference = 0.03Iteration = 10 Difference = 0.00Iteration = 11 Difference = 0.0002403903Iteration = 12 Difference = 0.0000084763Computing MP2 energy with 29 occupied and 66 virtual orbitals ... Transfering the 2e integrals from AO to MO: done 0%.Transfering the 2e integrals from AO to MO: done 10%.Transfering the 2e integrals from AO to MO: done 20%.Transfering the 2e integrals from AO to MO: done 30%.Transfering the 2e integrals from AO to MO: done 40%.Transfering the 2e integrals from AO to MO: done 50%.Transfering the 2e integrals from AO to MO: done 60%.Transfering the 2e integrals from AO to MO: done 70%.Transfering the 2e integrals from AO to MO: done 80%.Transfering the 2e integrals from AO to MO: done 90%.Energy=-171223.093981 MP2 Correlation Energy=-414.861844 Symmetry=C2V ENERGIES AND GRADIENT========== SCF RESULTS ==========Total Energy = -171223.0939809 (kcal/mol)Total Energy = -272.0 (a.u.)Electronic Kinetic Energy = 170947.5590687 (kcal/mol)Electronic Kinetic Energy = 272.2 (a.u.)The Virial (-V/T) = 2.0016eK, ee and eN Energy = -367133.0518992 (kcal/mol)Nuclear Repulsion Energy = 195909.9579183 (kcal/mol)======== POST SCF RESULTS ========MP2 Correlation Energy = -414.8618444 (kcal/mol)MP2 Correlation Energy = -0.0 (a.u.)Total Energy (with MP2 energy) = -171637.9558252 (kcal/mol)Total Energy (with MP2 energy) = -273.5224594 (a.u.)Occupied and Virtual Orbitals in MP2 = 29, 66========== SCF RESULTS ==========MOLECULAR POINT GROUPC2VEIGENV ALUES(eV)Symmetry: 1 B2 1 A1 2 B2 2 A1 3 A1 Eigenvalue: -303.674866 -303.674713 -303.631775 -303.630157 -303.627747 Symmetry: 3 B2 4 A1 5 A1 4 B2 6 A1 Eigenvalue: -303.623016 -303.612946 -30.261747 -29.204332 -27.434654 Symmetry: 5 B2 7 A1 8 A1 6 B2 1 B1 Eigenvalue: -25.107853 -22.681458 -21.177843 -21.160900 -17.947813 Symmetry: 1 A2 9 A1 7 B2 2 B1 8 B2 Eigenvalue: -16.982294 -16.493128 -16.444221 -15.528235 -15.090984 Symmetry: 10 A1 2 A2 11 A1 9 B2 3 B1 Eigenvalue: -14.549708 -13.900264 -13.511187 -12.866585 -12.585258 Symmetry: 12 A1 10 B2 3 A2 4 B1 13 A1 Eigenvalue: -12.306542 -12.249912 -11.875375 -11.724080 7.087684 Symmetry: 5 B1 11 B2 14 A1 15 A1 4 A2 Eigenvalue: 7.258967 7.333437 7.768019 8.007830 8.335639 Symmetry: 12 B2 16 A1 13 B2 6 B1 14 B2 Eigenvalue: 8.724559 9.351191 9.442577 9.514703 9.899670 Symmetry: 5 A2 7 B1 17 A1 6 A2 8 B1 Eigenvalue: 10.156153 10.202340 10.277209 10.321597 10.688224 Symmetry: 18 A1 19 A1 15 B2 16 B2 20 A1 Eigenvalue: 11.894808 12.191500 12.239221 13.969515 14.873546 Symmetry: 17 B2 18 B2 21 A1 9 B1 7 A2 Eigenvalue: 14.943412 24.174065 24.497423 25.322130 25.437565 Symmetry: 22 A1 10 B1 19 B2 23 A1 8 A2 Eigenvalue: 25.885952 26.483768 27.062307 27.959761 28.765862 Symmetry: 20 B2 24 A1 21 B2 25 A1 26 A1 Eigenvalue: 28.808775 29.825521 30.151628 30.719355 31.761393 Symmetry: 11 B1 22 B2 9 A2 23 B2 12 B1 Eigenvalue: 31.900881 32.902023 33.776600 33.784996 33.892372 Symmetry: 27 A1 13 B1 10 A2 28 A1 24 B2 Eigenvalue: 33.973347 35.433430 36.171829 36.518166 36.652103Symmetry: 14 B1 11 A2 29 A1 30 A1 15 B1 Eigenvalue: 36.898243 37.728039 37.748863 38.017197 38.224873 Symmetry: 25 B2 31 A1 26 B2 32 A1 27 B2 Eigenvalue: 38.257149 38.725616 38.931732 39.085808 40.204521 Symmetry: 12 A2 28 B2 16 B1 33 A1 29 B2 Eigenvalue: 41.526394 43.364548 49.700970 49.825069 55.635876 Symmetry: 34 A1 30 B2 35 A1 31 B2 36 A1 Eigenvalue: 60.068157 62.722443 64.506859 69.455078 77.091454 ATOMIC ORBITAL ELECTRON POPULATIONSC 1 S C 1 S C 1 Px C 1 Py C 1 Pz1.988309 0.353594 0.528720 0.555717 0.528189C 1 S C 1 Px C 1 Py C 1 Pz C 2 S1.112087 0.573252 0.345988 0.569138 1.989476C 2 S C 2 Px C 2 Py C 2 Pz C 2 S0.348915 0.547487 0.559715 0.527539 1.107065C 2 Px C 2 Py C 2 Pz C 3 S C 3 S0.400466 0.350437 0.582624 1.989604 0.349376C 3 Px C 3 Py C 3 Pz C 3 S C 3 Px0.546579 0.556665 0.525439 1.094947 0.398869C 3 Py C 3 Pz C 4 S C 4 S C 4 Px0.337608 0.578260 1.989672 0.349268 0.546835C 4 Py C 4 Pz C 4 S C 4 Px C 4 Py0.556721 0.525498 1.098025 0.397285 0.340660C 4 Pz C 5 S C 5 S C 5 Px C 5 Py0.579368 1.989603 0.349377 0.546403 0.556843C 5 Pz C 5 S C 5 Px C 5 Py C 5 Pz0.525439 1.094939 0.396197 0.340286 0.578257C 6 S C 6 S C 6 Px C 6 Py C 6 Pz1.989476 0.348915 0.551136 0.556063 0.527539C 6 S C 6 Px C 6 Py C 6 Pz C 7 S1.107065 0.406435 0.344470 0.582624 1.988310C 7 S C 7 Px C 7 Py C 7 Pz C 7 S0.353594 0.551270 0.533168 0.528188 1.112088C 7 Px C 7 Py C 7 Pz H 8 S H 8 S0.376075 0.543161 0.569141 0.455906 0.350817H 9 S H 9 S H 10 S H 10 S H 11 S0.455789 0.354839 0.455790 0.354841 0.454201H 11 S H 12 S H 12 S H 13 S H 13 S0.350338 0.454202 0.350338 0.453928 0.354491H 14 S H 14 S H 15 S H 15 S H 16 S0.453928 0.354490 0.453952 0.354422 0.453952H 16 S H 17 S H 17 S H 18 S H 18 S0.354420 0.453928 0.354491 0.453928 0.354491H 19 S H 19 S H 20 S H 20 S H 21 S0.454202 0.350338 0.454202 0.350338 0.455904H 21 S H 22 S H 22 S H 23 S H 23 S0.350817 0.455789 0.354840 0.455789 0.354840NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Mass (Mulliken) x y z1 6 -0.554993 1. -2. 0.00000244 12.011002 6 -0.413723 1. -1. 0.00000350 12.011003 6 -0.377347 -0. -0. 0.00000361 12.011004 6 -0.383332 -0. 0. 0.00000283 12.011005 6 -0.377344 -1. 1.08469892 -0.00000218 12.011006 6 -0.413724 -1. 2. 0.00000175 12.011007 6 -0.554995 -3.03550863 3.09049869 -0.00000256 12.011008 1 0.193277 2. -3. -0.00000215 1.008009 1 0.189371 0. -3. 0. 1.0080010 1 0.189370 0. -3. -0. 1.0080011 1 0.195461 1. -1. 0. 1.0080012 1 0.195460 1. -1. -0. 1.0080013 1 0.191581 -0. -1. 0. 1.0080014 1 0.191582 -0. -1. -0. 1.0080015 1 0.191626 0. 0. 0. 1.0080016 1 0.191628 0. 0. -0. 1.0080017 1 0.191581 -2. 0. -0. 1.0080018 1 0.191581 -2. 0. 0. 1.0080019 1 0.195461 -1. 2. 0. 1.0080020 1 0.195461 -1. 2. -0. 1.0080021 1 0.193278 -3.06212473 4. 0.00000114 1.0080022 1 0.189371 -3. 2. -0. 1.0080023 1 0.189371 -3. 2. 0. 1.00800Net Charge (Electrons):0.0000Dipole Moment (Debye):X: 0.0374 Y: 0.0265 Z: -0.0000 Ttl: 0.0458 Quadrupole Moment (Debye-Ang):XX: -50.1060 YY: -50.0433 ZZ: -49.0744XY: -0.0137 XZ: -0.0000 YZ: -0.0000Octapole Moment (Debye-Ang^2):XXX: 92.7823 YYY: -33.3817 ZZZ: -0.0002XYY: 30.1231 XXY: -13.8770 XXZ: -0.0000XZZ: 29.0156 YZZ: -12.3621 YYZ: -0.0000 XYZ: -0.0000 Hexadecapole Moment (Debye-Ang^3):XXXX: -913.8107 YYYY: -1448.3551 ZZZZ: -99.1216XXXY: 473.6560 XXXZ: -0.0005 YYYX: 469.1867YYYZ: 0.0004 ZZZX: -0.0003 ZZZY: 0.0004XXYY: -394.9777 XXZZ: -163.0937 YYZZ: -256.2102 XXYZ: 0.0002 YYXZ: -0.0002 ZZXY: 158.8551 HyperChem log stop -- Mon Dec 13 18:51:44 2010.。
HyperChem基本操作 Setup设置

HyperChem基本操作Setup设置Charge和Spin Multiplicity在Ab initio和Semi-empirical计算的对话框中出现。
Charge:指定额外的净剩电荷。
额外电荷定义当前的分子系统是一个电中性系统,正电系统(阳离子),还是一个负电系统(阴离子)。
Spin Multiplicity:自旋多重度。
闭壳分子的自旋多重度为1(单重态)。
一个自由基,有一个未成对电子,自旋多重度为2(双重态)。
有两个未成对电子的系统,自旋多重度一般为3(三重态)。
然而在某些情况下,例如两个自由基,两个未成对电子也可能产生单态。
State描述系统中价电子的状态。
包括指定分子处于第一激发单态(Next lowest)或者Lowest电子态。
Lowest给定自旋多重度的最低电子态。
它不一定是基态。
Next lowest给定自旋多重度(单重、双重、三重或四重态)的第一电子激发态。
在HyperChem 6.0中,semi-empirical方法通常需要给定多重度的最低能态(lowest)或者次最低能态(next lowest)的计算。
由于偶数个电子的分子没有未成对电子,是闭壳层单态,所以只有最低三重态是有效的,而次最低三重态是无效的。
例如,苯有偶数个电子,并且基态是闭壳层单重态。
我们可以计算基态(最低单重态),第一激发单重态(次最低单重态),或者第一激发三重态(最低三重态)。
也就是说,或者HOMO被两个电子占据,或者一个电子在HOMO,另一个电子在LUMO,产生了激发单重态或者三重态。
对双重态和四重态,只有给定多重度的最低态(lowest)可用。
UHF选项仅允许给定多重度的最低态(lowest)可用。
例如,可以用UHF选项研究苯的最低三重激发态,但是不能用来计算单重激发态。
这是因为HyperChem中的UHF选项不允许任意的轨道占据,也不允许CI计算。
对于RHF选项,可以计算CI波函数。
这个计算由一系列计算得到的RHF轨道开始,或者从最低单态(或双重态),或者从half electron的单态和三态(或双态和四重态)轨道。
HyperChem

分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
HyperChem
分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
谷晓明 物理化学HyperChem
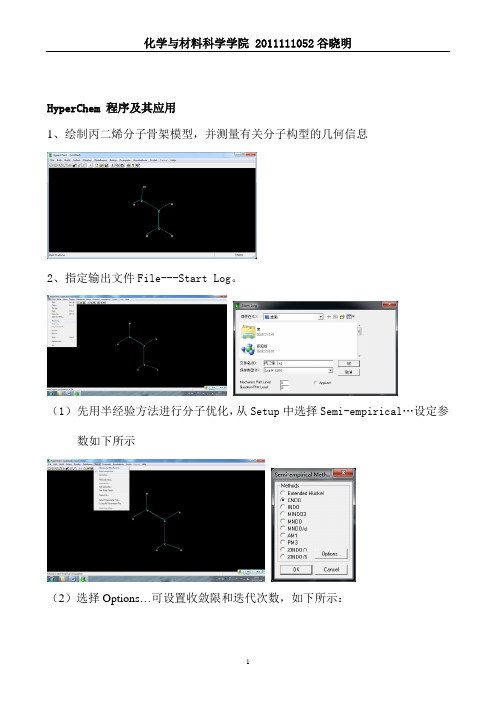
HyperChem 程序及其应用1、绘制丙二烯分子骨架模型,并测量有关分子构型的几何信息2、指定输出文件File---Start Log。
(1)先用半经验方法进行分子优化,从Setup中选择Semi-empirical…设定参数如下所示(2)选择Options…可设置收敛限和迭代次数,如下所示:(3)从Compute中选择Geometry Optimzation…进行集合构型优化:(4)优化完成之后,在Compute选择Single Point可进行单点计算。
3、采用从头算的方法:(1)Setup中选择Ab Initio…设定参数如下:(2)从Compute中选择Geometry Optimzation…进行集合构型优化:(3)完成集合构型优化后,从Compute选择Single Point可进行单点计算。
4、计算结束后,停止数据输出,从File---Stop Log。
5、分析有关分子的性质并简单分析讨论分子性质(1)采用从头算方法后,分析振动光谱:(该图显示谱线的位置、强度和振动模式)虚振动频率-185.84意味着,此结构不是一个稳定结构,而是一个过渡态。
(2)计算电子光谱最低能量跃迁π-π*在373.90,是禁阻跃迁允许的跃迁是116.84单态π-π*跃迁。
(3)分子偶极矩(4)轨道特征1、最高占据轨道2、最低空轨道(5)绘分子图,测电子光谱从Comput选择Plot Molecular Graphs1、2D图像2、3D图像6、结论与经验1、丙烯分子为一平面型分子,并且其振动频率存在虚频-185.84,意味着此平面结构不是一个稳定结构,而是一个过渡态。
2、半经验算法计算分子总能量为-16180.6852898 (kcal/mol),从头算方法计算分子总能量为-72576.4084722 (kcal/mol),所以计算方法的选择很重要。
3、计算分子的电子光谱能够得到该分子最低能量跃迁π-π*在373.90,是禁阻跃迁;允许的跃迁是116.84单态π-π*跃迁。
hyperchem教案3

一、建立H2O2模型双击Draw—选择O原子---双击选择图标---选择H1-O1-O2-H2四个原子----edit---set bond torsion ----bond torsion=60度---OK---save as 到指定文件夹(用英语或汉语拼音命名。
扩展名为.hin)二、优化H2O2的结构1、先进行MM+优化File----start log----命名H2O2----append----OKSetup----molecular mechanics----MM+---OK----OKComput---optimization----OKFile----stop log2、进行semi-empirical优化File---start log---OKSetup----semi-empirical----AM1---OKComput---optimization----OKFile----stop log3、进行ab-initio优化File---start log---OKSetup----ab-initio---将basis set 设置为6-31g*---OKComput---optimization----OKFile----stop log三、进行单点能计算、振动光谱(IR)计算和电子光谱(UV)计算1、单点能计算File---start log---OKSetup----ab-initio---将basis set 设置为6-31g*---OKComput---single point----OKFile----stop log2振动光谱(IR)File---start log---OKSetup----ab-initio---将basis set 设置为6-31g*---OKComput---vibrations----OKFile----stop log3、电子光谱(UV)计算File---start log---OKSetup----ab-initio---将basis set 设置为6-31g*---options---configration intereaction—singly exicited—将energy criterion 设置为100eVeV—OKComput---single point----OKFile----stop log三、输出和保存结果File----export----勾选有关项后---OK----命名为*****.ext 文件----四、观察和输出体系能量、原子电荷、键长、键级、键角、HOMO、LUMO、分子静电势、IR、UV在display----lable中可看到原子电荷、键长、键级、键角在comput的中部可以看到体系能量、HOMO、LUMO、分子静电势、IR、UV. (copy----paste)。
HyperChem应用-水分子轨道计算

HyperChem应用水分子轨道计算构造水分子1. 在Display菜单,确保Show Hydrogens打开,Rendering对话框Sticks栏中的Perspe ctive关闭。
2. 关闭Default Element对话框中的Explicit Hydrogens,设置缺省元素为O。
3. 在工作区画一个氧原子。
4. 双击Selection激活Model Builder。
它自动为氧原子添加氢原子。
5. 设置Label添加元素符号。
结果是这样:∣H∣∣∣O╲╲╲H键角是109度。
结构校准1. 在Edit菜单选择Align Molecules。
2. 在Align对话框选择Secondary,With对话框选择Y Axis。
3. 关闭Minor Axis。
4. 选择OK。
得到这样的水分子:H H╲╱╲╱╲╱╲╱O5. 把这个结构保存为h2o.hin。
显示原子电荷1. 打开Labels对话框。
2. 把Charge作为Atom的选项,单击OK。
计算波函1. 在Setup菜单选择Semi-empirical。
2. 选择CNDO方法。
选择Options。
当然也可以选择其他方法。
3. 在Semi-empirical的Options的对话框中使用下面的值:Total charge: 0, Spin multiplicity: 1, Spin Pairing: RHF,State: Lowest, Convergence limit: 0.0001, Iteration limit: 50,Accelerate convergence: NO。
这意味着两次叠代计算的值的差小于0.0001 kcal/mol,叠代次数已经达到了最大的次数5 0次。
4. 选择OK回到工作区。
5. 选择Compute菜单中的Single Point。
结果:能量,梯度,和原子电荷如图所示:0.145 0.145╲╱╲╱╲╱╲╱-0.290窗口左下角:Energy=-320.414117, Gradient=124.385845, Symmetry=C2V(结果可能会有略微差别。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
*保存文件
• File→Save As→
保存←
*改变键型连接
• 将鼠标移动至需要修改键型的原子上面,左击鼠标同时移 动至另一个原子上面,则
*制作苯环
• 用鼠标单击环形结构中的任一原子,则可画出苯环
*标记原子
• Display→Labels ↓ →→Symbol→OK ↑ ↓ ↑ ↑ →→↑
*编辑个人原子
Ala为丙氨酸 Val为缬氨酸 Gly为甘氨酸
构造• • • •
进入Amino Acids选择Beta Sheet 选择两个以上氨基酸建立肽链 关闭Amino Acids Display→Labels→确认Atoms和Redidues选择的是 None→OK
*创建一个两性分子
Hyperchem学习
小组成员:
胡非非 王珊珊 张鹏程 杨素 邓超 张春洪 花木兴 李庆湖 龙继亮
概况:
• HyperChem用户界面的介绍 • 讨论如何使用工具、菜单和菜单项建立、编辑、显示和操 纵分子
学习内容:
• 打开——打开、关闭某一文件,使用标签和分子效果图 • 基本绘图和编辑——画图、选择、复制及删除原子连接 • 创建小分子的2D和3D模型——绘制和编辑2D图将其转 为3D模型 • 交换、旋转、缩放分子——移动和操纵分子 • 测量结构属性——测量和调整分子几何结构 • 创建一个多肽——创造生物聚合物,学会使用氨基酸和 酸数据库
*Z轴的剪辑
双击
意移 观动 察两 变侧 化红 框 部 分 注
OK
五、测量结果属性
• • • • • • 创建2D结构 编辑此结构 建立3D框架 查看某个原子 键长 键角测量
*创建2D结构
*编辑此结构
双击环状结构
Build
Default Element
N
将一个 碳原子 换成氧原子, 同时点击 DISPLAY 接着 点击Labers 选 择Symbol
四、交换、旋转、缩放分子
• • • • • • • XY轴工具的使用 Z轴工具的使用 缩放工具的使用 定心和缩放 XY轴旋转 Z轴旋转 Z轴剪辑
*XY轴工具的使用
• →工作区→移动鼠标,则所画图像随着移动→双击 ↓
输入XY轴的变化量 如: ↓ OK
←
*Z轴工具的使用
• Display→Rendering→ →Vector and
*多原子的选择
• Select→Turn off Select Sphere→同时点击鼠标的左右键, 圈出想要的多个原子→Select→Turn on Multiple Selections→同时点击鼠标的左右键→放开鼠标
*删除原子
• 将鼠标移动至
点击左键
• 选择所要删除的原子或者键型右击鼠标
*复制原子到Clipboard
• 双击鼠标左键→Element Table→选择原子→关闭Element Table→将鼠标移动至想要编辑的原子上面,单击左键, 则该原子变为自己后面所选原子,如
*选用模型生成器
• Build→Add H and Model Build→
如果没有显示相应原子,则进入 Display→Show Hydrogens
原子间距离显示在 状态栏
*键角测量
• 按住鼠标左键从碳原子拖到氢原子,接着放开鼠标则
键角显示
六、创建多肽
• • • •
Amino Acids对话框的使用 创建一个多肽 创建一个两性分子 位点专一诱变
*Amino Acids对话框的使用
• Databases→Amino Acids→
不同氨基酸结构
OK Thr Mutate
谢谢观赏
胡非非
hyperchem学习 hyperchem
Line Options→ 移动鼠标 ← ↓ ←
→Turn on Perspective ↓ ←确定
*缩放工具的使用
按着鼠标左键移 动鼠标
*定心和缩放
• Display Select to Fit
恢复原来大小
*XY轴转换
移动鼠标
可进行XY轴的旋转
*Z轴的旋转
将鼠标移动至工作区, 则可以对分子进行旋转
*画原子
Build→Default Element→
↓ ← Properties Ok←
*画原子
• Ok→工作区→左键→
*连接
• 将鼠标移动至某一画好的碳原子上,按住鼠标将其拖至另 一相连的碳原子上边,放开鼠标,则:
*选择原子
Select→Atoms→Turn off Multiple Selections→ ↓ 将鼠标移动至想要的原 子或者其他原子上面则 可对相应原子进行选择
• Edit→Rotate→ →
调整X Y Z轴 的数据点击 OK
Make Zwitterion Databases
*位点专一诱变
• Display→Labels→在Residues里选择Name+Seq→OK
Select
Residues
接 下 页
*位点专一诱变
接上图 右击鼠标 点击GLY3
Databases
*打开文件
• File→Open→Hyperchem→Samples→c60.hin→
*使用laber
• Display→Labels→
*使用不同Moleculer Renderings
• Display→Rendering→
↓ ←
Balls
一、打开
二、基本绘图和编辑
• • • • • • Drawing Atoms Drawing Bonds Selecting Atoms Selecting Groups of Atoms Deleting Atoms Copying Atoms to the Clipboard
• 将鼠标移动至
点击左键
• 将鼠标移动至某一原子或键型,点击左键
三、小分子的2D和3D模型
1,画2D图 2,保存文件 3,改变键型连接 4,制作苯环 5,标记原子 6,编辑个人原子 7,调用模型生成器
*画2D图
• Build→Turn on Arbitray Valence→Turn off Explicit Hydrogens→Default Element→Element Table→选择原 子→ →画图如下
*建立3D框架
• Build→Turn off Explicit Hydrogens→双击
*查看某个原子
• Select→关闭Atoms和Multiple Seltctions→
点击氧原子
注意此处
*键长
• 单击碳氧键
键长表示在 此处
*键长测量
• Select→Turn on Multiple Selections→右击鼠标(选中所 有原子)→左击鼠标两个原子→