HyperChem 程序及其应用
化学信息学——第八讲hyperchem 软件学习

HyperChem程序简介 7.5版新增功能 OpenGL绘图: 基础演示模式:已全部转换为全新的 OpenGL模式,使分子演示图形具有更高 质量支持自定义色彩,1600万种颜色代替 了传统的8种标准色。 混合演示:棒型,球型或组合型等分子 演示手段可以用于该分子的任意原子及链 段的演示,及全新的适用于原子的"管型" 演示。增强的蛋白质模型构建功能,支持 多种蛋白质二级结构演示,大分子的电子 密度近似方法。
13
工具 条 •使用图标工具可以简化操作 •从左至右分别是绘制、选择、旋转、平面 转动、平移
14
工作状态
•工作 区: •状态 行:
15
2、打开已存在的数据文件 •File-Open
16
浏览文件夹
17
C60分子
18
选择分子图形的显示方式 •Display- Labels可以选择原子、化学键等 标记方式:
39
计算几何构型 •COMPUTE---GEOMETRY
40
计算结果 •优化结束
41
测量键长、键角、二面角等结构参数 •数值显示在底部状态栏
42
单点能计算
•对给定体系优化构型后,可对优化后的构 型再进行单点计算。
43
单点能结果 •单点能结果
44
输出结果
45
计算结果的输出文件
• • • • • • • • • • • • • • • HyperChemlog start -- Sun May 28 21:38:15 2000. Geometry optimization, SemiEmpirical, molecule = (untitled). CNDO PolakRibiere optimizer Convergence limit = 0.0001000 Iteration limit = 50 Accelerate convergence = YES Optimization algorithm = Polak-Ribiere Criterion of RMS gradient = 0.0100 kcal/(A mol) Maximum cycles = 150 RHF Calculation: Singlet state calculation Number of electrons = 22 Number of Double Occupied Levels = 11 Charge on the System = 0 Total Orbitals = 22
HyperChem

分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
HyperChem
分子图形与分子模型设计——HyperChem使用简介厦门大学化学系2005年3月HyperChem使用简介HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。
HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。
它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。
HyperChem 7.5版本已经推出。
图1是HyperChem的工作窗口,最下部是工作状态档。
在菜单下面是常用工具档。
图1 HyperChem的工作窗口HyperChem的操作可以使用鼠标和键盘两种。
在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状:1.绘图工具。
鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。
2.选择工具。
3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。
4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。
5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。
6.z轴平移工具。
7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。
8.z轴截片工具。
鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。
一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。
HyperChem的详细操作将结合具体的实例进行讲解。
以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。
一、File1.New (Ctrl+N):新建一个沿尚未命名文件。
谷晓明 物理化学HyperChem
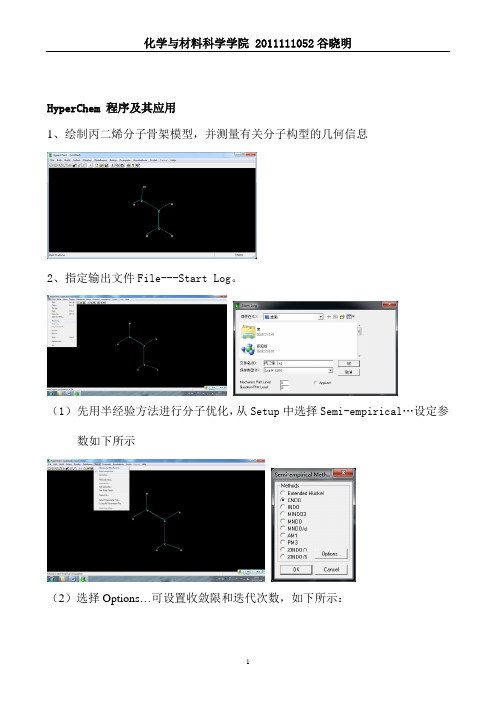
HyperChem 程序及其应用1、绘制丙二烯分子骨架模型,并测量有关分子构型的几何信息2、指定输出文件File---Start Log。
(1)先用半经验方法进行分子优化,从Setup中选择Semi-empirical…设定参数如下所示(2)选择Options…可设置收敛限和迭代次数,如下所示:(3)从Compute中选择Geometry Optimzation…进行集合构型优化:(4)优化完成之后,在Compute选择Single Point可进行单点计算。
3、采用从头算的方法:(1)Setup中选择Ab Initio…设定参数如下:(2)从Compute中选择Geometry Optimzation…进行集合构型优化:(3)完成集合构型优化后,从Compute选择Single Point可进行单点计算。
4、计算结束后,停止数据输出,从File---Stop Log。
5、分析有关分子的性质并简单分析讨论分子性质(1)采用从头算方法后,分析振动光谱:(该图显示谱线的位置、强度和振动模式)虚振动频率-185.84意味着,此结构不是一个稳定结构,而是一个过渡态。
(2)计算电子光谱最低能量跃迁π-π*在373.90,是禁阻跃迁允许的跃迁是116.84单态π-π*跃迁。
(3)分子偶极矩(4)轨道特征1、最高占据轨道2、最低空轨道(5)绘分子图,测电子光谱从Comput选择Plot Molecular Graphs1、2D图像2、3D图像6、结论与经验1、丙烯分子为一平面型分子,并且其振动频率存在虚频-185.84,意味着此平面结构不是一个稳定结构,而是一个过渡态。
2、半经验算法计算分子总能量为-16180.6852898 (kcal/mol),从头算方法计算分子总能量为-72576.4084722 (kcal/mol),所以计算方法的选择很重要。
3、计算分子的电子光谱能够得到该分子最低能量跃迁π-π*在373.90,是禁阻跃迁;允许的跃迁是116.84单态π-π*跃迁。
HyperChem程序及其应用
河北师范大学计算量子化学研究所蔡新华教授量子化学在线教学.100/qc/lzhx-0.htmHyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作区:状态行:2、打开已存在的数据文件File-Open选择分子图形的显示方式Display- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
HyperChem应用-水分子轨道计算

HyperChem应用水分子轨道计算构造水分子1. 在Display菜单,确保Show Hydrogens打开,Rendering对话框Sticks栏中的Perspe ctive关闭。
2. 关闭Default Element对话框中的Explicit Hydrogens,设置缺省元素为O。
3. 在工作区画一个氧原子。
4. 双击Selection激活Model Builder。
它自动为氧原子添加氢原子。
5. 设置Label添加元素符号。
结果是这样:∣H∣∣∣O╲╲╲H键角是109度。
结构校准1. 在Edit菜单选择Align Molecules。
2. 在Align对话框选择Secondary,With对话框选择Y Axis。
3. 关闭Minor Axis。
4. 选择OK。
得到这样的水分子:H H╲╱╲╱╲╱╲╱O5. 把这个结构保存为h2o.hin。
显示原子电荷1. 打开Labels对话框。
2. 把Charge作为Atom的选项,单击OK。
计算波函1. 在Setup菜单选择Semi-empirical。
2. 选择CNDO方法。
选择Options。
当然也可以选择其他方法。
3. 在Semi-empirical的Options的对话框中使用下面的值:Total charge: 0, Spin multiplicity: 1, Spin Pairing: RHF,State: Lowest, Convergence limit: 0.0001, Iteration limit: 50,Accelerate convergence: NO。
这意味着两次叠代计算的值的差小于0.0001 kcal/mol,叠代次数已经达到了最大的次数5 0次。
4. 选择OK回到工作区。
5. 选择Compute菜单中的Single Point。
结果:能量,梯度,和原子电荷如图所示:0.145 0.145╲╱╲╱╲╱╲╱-0.290窗口左下角:Energy=-320.414117, Gradient=124.385845, Symmetry=C2V(结果可能会有略微差别。
HyperChem程序及其应用
河北师范大学计算量子化学研究所蔡新华教授量子化学在线教学.100/qc/lzhx-0.htmHyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作区:状态行:2、打开已存在的数据文件File-Open选择分子图形的显示方式Display- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
HyperChem基本操作 单点计算

HyperChem基本操作单点计算单点计算,仅仅执行势能曲面上的一个单个点的计算。
例如对一个双原子分子来说,这可能是在点a原子间距离R=2.0埃的计算。
单点计算的结果给出系统在当前几何构型的势能,以及那个点的梯度。
在点b,c,d,或e的单点计算可能将给出更高的能量。
如果在关键的部分取足够多的点,利用Origin或者Matlab等数学工具软件,就能描出势能曲线,从而精确算出离解能De和核平衡距离re,利用公式就能得到震动参数ωe。
对多原子系统来说,状况更加复杂,但是本质是一样的。
Orbital中显示的轨道信息Alpha & Beta显示选择的轨道是alpha自旋还是beta自旋。
LUMO+显示选择轨道相对于LUMO的位置关系。
例如,选择能量在LUMO之上的轨道,文本框依次显示+1,+2,+3......;如果选择能量低于LUMO的轨道,文本框显示-1,-2,-3......。
HOMO-显示选择轨道相对于HOMO的位置关系。
例如,选择能量在HOMO之上的轨道,文本框依次显示-1,-2,-3......;如果选择能量低于HOMO的轨道,文本框显示+1,+2,+3......。
Number显示从能量最低轨道开始的选择轨道绝对数值。
对于UHF计算,轨道的alpha和beta列编号分开显示,对HOMO-和LUMO+选项也是这样。
Energy以eV为单位显示选择轨道的能量。
Symmetry显示选择轨道的不可约表示。
Labels在轨道显示窗口中,显示每个轨道的电子占据情况和每个轨道的能量。
按照约定俗成,向上的箭头代表alpha自旋,向下的箭头代表beta自旋。
对于RHF计算,最大轨道占据是2;对于UHF计算,最大轨道占据是1。
Zoom Out和轨道的放大如果轨道间距太密,在使用Labels时,文字符号会重合在一起,看起来很不方便。
通过鼠标左键圈出一个或相邻几个选择的轨道,这些轨道就会被放大。
放大之后,Zoom Out选项可以在窗口中重新回到显示全部轨道范围。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
HyperChem 程序及其应用一、HyperChem 程序的运行环境HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。
可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。
该公司的网址为:该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。
1、软件环境Windows95、Windows98或Windows2000系统。
2、硬件环境486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。
为了能够以最佳方式显示分子图像,最好有VGA以上显示器。
3、程序安装使用该程序应注意程序版权(注册)。
安装程序默认子目录为:C:\hyper6安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。
有关该程序的使用说明、参考手册等全套文档均可免费获得。
二、程序基本使用方法我们以演示版本为例,说明该程序的基本使用方法。
1、启动程序在屏幕上,双击绿色烧杯可以得到如下画面:点击 Try进入工作区窗口窗口各部分功能简介标题名称:最大、最小化、退出按钮菜单条:FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、SETUP 、COMPUTE、CANCEL、SCRIPT、HELP工具条:工作 区:状态 行:2、打开已存在的数据文件 File-OpenDisplay- Labels可以选择原子、化学键等标记方式:Dispay-RenderingRenderings-BallsRendering—Balls and Cylinders3、建立计算分子的数据文件以丁二烯为例:选择Build-Default Element可以显示指定元素的基本性质:选择绘图工具后,得到碳碳骨架。
左键、右键增减键序把2D骨架转换为3D图像Select选定原子后,状态行提示原子标号、坐标等结构信息。
保存为数据文件如需要修改原子骨架,比如把某个氢原子改为氯原子: 选定缺省元素后,在指定原子位置上双击即可。
移动、旋转、放大、缩小分子图像此时,可以用Build—Set设定初始数据,如键长、键角、形式电荷等等。
也可以用来直接测定分子构型数据三、优化分子构型1、丁二烯分子体系能量极小值的计算记录输出结果: FILE---START LOG….绘出丁二烯分子骨架模型:DRAW测量键长、键角、二面角等结构信息:SELECT用半经验方法优化分子构型 SETUP--CNDOCOMPUTE---GEOMETRY测量键长、键角、二面角等结构参数。
对给定体系优化构型后,可对优化后的构型再进行单点计算。
计算结果的输出文件HyperChem log start -- Sun May 28 21:38:15 2000.Geometry optimization, SemiEmpirical, molecule = (untitled).CNDOPolakRibiere optimizerConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESOptimization algorithm = Polak-RibiereCriterion of RMS gradient = 0.0100 kcal/(A mol) Maximum cycles = 150RHF Calculation:Singlet state calculationNumber of electrons = 22Number of Double Occupied Levels = 11Charge on the System = 0Total Orbitals = 22Starting CNDO calculation with 22 orbitalsE=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=7144.22066]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=2 Diff=5.36263]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=3 Diff=0.48300]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=4 Diff=0.05233]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=5 Diff=0.00750]E=0.0000 Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=6 Diff=0.00009]E=-2659.4619 Grad=53.592 Conv=NO(0 cycles 1 points) [Iter=1 Diff=4.99709] E=-2659.4619 Grad=53.592 Conv=NO(0 cycles 1 points) [Iter=2 Diff=0.38262] E=-2671.4854 Grad=0.022 Conv=NO(26 cycles 68 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.042 Conv=NO(26 cycles 69 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.033 Conv=NO(27 cycles 70 points) [Iter=1 Diff=0.00001] E=-2671.4854 Grad=0.019 Conv=NO(27 cycles 71 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.015 Conv=NO(28 cycles 72 points) [Iter=1 Diff=0.00001] E=-2671.4854 Grad=0.069 Conv=NO(28 cycles 73 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.011 Conv=NO(29 cycles 74 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.012 Conv=NO(29 cycles 75 points) [Iter=1 Diff=0.00000]E=-2671.4854 Grad=0.012 Conv=NO(30 cycles 76 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.009 Conv=NO(30 cycles 77 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.010 Conv=NO(30 cycles 78 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.022 Conv=NO(30 cycles 79 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.011 Conv=NO(31 cycles 80 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.017 Conv=NO(31 cycles 81 points) [Iter=1 Diff=0.00000] E=-2671.4854 Grad=0.006 Conv=YES(32 cycles 82 points) [Iter=1 Diff=0.00000]ENERGIES AND GRADIENTTotal Energy = -20550.8323392 (kcal/mol)Total Energy = -32.749182076 (a.u.)Binding Energy = -2671.4854530 (kcal/mol)Isolated Atomic Energy = -17879.3468863 (kcal/mol)Electronic Energy = -55625.1226392 (kcal/mol)Core-Core Interaction = 35074.2903000 (kcal/mol)Heat of Formation = -1675.3134530 (kcal/mol)Gradient = 0.0062527 (kcal/mol/Ang)MOLECULAR POINT GROUPC2HEIGENVALUES(eV)Symmetry: 1 AG 1 BU 2 AG 2 BU 3 AG Eigenvalue: -43.673668 -37.506996 -29.111160 -28.857952 -23.531034 Symmetry: 3 BU 1 AU 4 BU 4 AG 5 AG Eigenvalue: -22.587774 -20.271618 -16.989029 -16.868931 -14.247993 Symmetry: 1 BG 2 AU 5 BU 6 AG 6 BU Eigenvalue: -13.322699 3.356511 6.598820 7.283803 7.489040 Symmetry: 2 BG 7 AG 7 BU 8 AG 8 BU Eigenvalue: 7.776278 8.189266 8.610060 10.810771 11.913039 Symmetry: 9 AG 9 BUEigenvalue: 13.934214 14.264333ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C 1.059899 0.950186 1.006745 1.012745 1.003647AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C 0.973639 1.009929 0.987255 1.003647 0.973639AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C 1.009929 0.987255 1.059899 0.950186 1.006745AO: 4 Pz C 5 S H 6 S H 7 S H 8 S H 1.012745 0.997504 0.993683 1.004769 1.004769AO: 9 S H 10 S H0.997504 0.993683NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 -0.029575 0.13401 -1.42160 0.00000 12.011002 6 0.025530 0.08094 -0.09964 -0.00000 12.011003 6 0.025530 -1.09703 0.72952 0.00000 12.011004 6 -0.029575 -1.15010 2.05149 -0.00000 12.011005 1 0.002496 1.07933 -2.00856 -0.00000 1.008006 1 0.006317 -0.75684 -2.08833 -0.00000 1.008007 1 -0.004769 1.05152 0.45978 0.00000 1.008008 1 -0.004769 -2.06762 0.17011 -0.00000 1.008009 1 0.002496 -2.09542 2.63844 0.00000 1.0080010 1 0.006317 -0.25925 2.71822 0.00000 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 0.00121 -0.01081 0.000122 6 0.00599 0.01255 -0.000213 6 -0.00607 -0.01263 0.000224 6 -0.00120 0.01073 -0.000125 1 0.00859 -0.00497 -0.000026 1 -0.00946 -0.00870 -0.000037 1 0.00169 0.00240 0.000048 1 -0.00161 -0.00235 -0.000059 1 -0.00867 0.00502 0.0000210 1 0.00953 0.00875 0.00003Dipole (Debyes) x y z TotalPoint-Chg. -0.000 0.000 0.000 0.000sp Hybrid -0.000 0.000 0.000 0.000pd Hybrid 0.000 0.000 0.000 0.000Sum -0.000 0.000 0.000 0.000Single Point, SemiEmpirical, molecule = (untitled).CNDOConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESSingle Point, SemiEmpirical, molecule = (untitled).CNDOConvergence limit = 0.0001000 Iteration limit = 50Accelerate convergence = YESRHF Calculation:Singlet state calculationNumber of electrons = 22Number of Double Occupied Levels = 11Charge on the System = 0Total Orbitals = 22Starting CNDO calculation with 22 orbitalsIteration = 1 Difference = 7100.34970Iteration = 2 Difference = 5.91366Iteration = 3 Difference = 0.55648Iteration = 4 Difference = 0.06489Iteration = 5 Difference = 0.00841Iteration = 6 Difference = 0.00014Iteration = 7 Difference = 0.00001Energy=-2671.485453 Gradient=0.006301 Symmetry=C2HENERGIES AND GRADIENTTotal Energy = -20550.8323392 (kcal/mol)Total Energy = -32.749182076 (a.u.)Binding Energy = -2671.4854529 (kcal/mol)Isolated Atomic Energy = -17879.3468863 (kcal/mol) Electronic Energy = -55625.1216733 (kcal/mol)Core-Core Interaction = 35074.2893341 (kcal/mol)Heat of Formation = -1675.3134529 (kcal/mol) Gradient = 0.0063010 (kcal/mol/Ang)MOLECULAR POINT GROUPC2HEIGENVALUES(eV)Symmetry: 1 AG 1 BU 2 AG 2 BU 3 AG Eigenvalue: -43.673672 -37.507000 -29.111160 -28.857954 -23.531034Symmetry: 3 BU 1 AU 4 BU 4 AG 5 AG Eigenvalue: -22.587776 -20.271622 -16.989029 -16.868935 -14.247996Symmetry: 1 BG 2 AU 5 BU 6 AG 6 BU Eigenvalue: -13.322702 3.356504 6.598815 7.283800 7.489039Symmetry: 2 BG 7 AG 7 BU 8 AG 8 BU Eigenvalue: 7.776274 8.189266 8.610056 10.810770 11.913034Symmetry: 9 AG 9 BUEigenvalue: 13.934207 14.264326ATOMIC ORBITAL ELECTRON POPULATIONSAO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C 1.059899 0.950186 1.006744 1.012745 1.003647AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C 0.973639 1.009930 0.987255 1.003647 0.973639AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C 1.009930 0.987254 1.059899 0.950186 1.006744AO: 4 Pz C 5 S H 6 S H 7 S H 8 S H 1.012746 0.997504 0.993683 1.004769 1.004769AO: 9 S H 10 S H0.997503 0.993682NET CHARGES AND COORDINATESAtom Z Charge Coordinates(Angstrom) Massx y z1 6 -0.029575 0.13401 -1.42160 0.00000 12.011002 6 0.025530 0.08094 -0.09964 -0.00000 12.011003 6 0.025530 -1.09703 0.72952 0.00000 12.011004 6 -0.029575 -1.15010 2.05149 -0.00000 12.011005 1 0.002496 1.07933 -2.00856 -0.00000 1.008006 1 0.006317 -0.75684 -2.08833 -0.00000 1.008007 1 -0.004769 1.05152 0.45978 0.00000 1.008008 1 -0.004769 -2.06762 0.17011 -0.00000 1.008009 1 0.002497 -2.09542 2.63844 0.00000 1.0080010 1 0.006318 -0.25925 2.71822 0.00000 1.00800ATOMIC GRADIENTSAtom Z Gradients(kcal/mol/Angstrom)x y z1 6 0.00089 -0.01031 0.000152 6 0.00652 0.01230 -0.000253 6 -0.00671 -0.01284 0.000254 6 -0.00129 0.01086 -0.000095 1 0.00889 -0.00516 -0.000066 1 -0.00948 -0.00880 -0.000037 1 0.00169 0.00244 0.000048 1 -0.00152 -0.00235 -0.000069 1 -0.00861 0.00493 -0.0000010 1 0.00963 0.00894 0.00005Dipole (Debyes) x y z Total Point-Chg. -0.000 0.000 0.000 0.000 sp Hybrid -0.000 -0.000 0.000 0.000 pd Hybrid 0.000 0.000 0.000 0.000 Sum -0.000 0.000 0.000 0.0002、二氧化碳分子例用分子力学方法优化分子结构用半经验方法CNDO 优化分子结构选择优化方法和优化收敛限。