临床试验启动会议纪要
药物临床试验之研究者会议 ppt

研究者会议
-
1
研究者会议
研究者会议是试验过程中必不可少的部分。 在中国GCP中第六十六条中明确规定了: “对于多中心试验组织实施要在临床试验 开始前及进行的中期应组织研究者会 议……”,可见研究者会议的召开也是法 规的要求。
研究者会议Investigator meeting(常用缩写 IM),或“方案讨论-会”,主要是各参研 2
会议日程 签到表 会议纪要 会议材料/PPT Q&A
-
5
总之,研究者会议我们除开要完善会场布置之外,尽 量把工作重点往方案敲定、培训操作和讨论、统一思 想和项目动员这几个方面靠拢,如果做到了,我们研 究者会议就基本达到了。
-
6
谢谢
-
7
目的
对方案和临床试验操作流程进行讨论 确保在试验过程中的可操作性 对可能出现的试验问题进行预估和防范 整个项目团队首次见面,彼此熟悉的过程 对整个项目团队进行首次全面的培训
-
3
内容
临床专业领域知识
研究背景
方案介绍研究药物介绍来自安全性管理数据管理
中心实验室操作管理-
4
文件处理归档
临床试验研究者告知书(11版)

临床试验研究者告知书(1.1版)尊敬的研究者:为确保整个项目顺利实施,我们将项目实施过程中的注意事项向您告知:1.为了确保项目顺利启动,建议您在项目实施过程中有任何疑问,可与机构办人员及时联系,我们将不遗余力为您协调处理。
2.项目启动前,由专业组负责人确认主要研究者人选名单并向机构办公室(1)备案,如无特殊原因,由主要研究者组建临床研究小组,确定研究成员名单。
监查员或机构办公室人员将项目相关资料(附件一)递交给您,请您审核方案等,并协助项目负责人填写《伦理申请表》(附件二),递交机构办公室相关责任人处(2或3)。
3.待伦理委员会出审查结果出具批件后,协助机构办公室(2或3)完成项目检查费报价单(以EMAIL形式确认)。
4.项目合同签署时,请务必打印“临床试验合同签署意见表”(中心章,审批时限1个月;机构章,审批时限2周,附件三),主要研究者签署确认后将合同一并递交机构办公室(2或3);同样,在签署总结报告时,也需打印“临床试验总结报告签署意见表”(附件四);相关的申请表格最新内容我们均已公布在我中心内网GCP专栏内。
5.协议签订后,由主要研究者与申办者共同确定启动会日期并向机构办公室报备(2或3),由主要研究者或机构办公室发布会议启动通知。
召开启动会时,应指定一名临床试验协调员/秘书负责记录并撰写会议纪要,方案及可行性讨论后,由主要研究者明确分工授权(参见附件五);6.临床试验协调员/秘书保存启动会相关资料(原件,签到表、PPT和会议纪要等)。
机构办公室(2或3)负责文档归档的监督,并协同主要研究者进行中心内外的协调。
7.项目实施前,药物送至金山总部或市区分部专业组试验药物存放室。
试验药物到达前必须实现与我办公室秘书(2)联系,确保药物是由三方共同签字确认(申办方、药物管理员、机构办公室);同样,在回收药物至申办方时,也请实现联系机构办公室秘书(2)到场确认签字。
8.项目实施中,请PI任命项目组质控员,项目质控员完成项目组内审(两月1次),检查问题上报PI进行项目组内部整改,PI对项目的实施质量负责;专业组质控员每半年度完成专业组内审(每半年一次,集中在4月和10月),质量检查结果进行整改并上报专业组负责人。
临床试验项目启动会SOP

临床试验项目启动会SOP
I目的规范临床试验项目启动会规程,保证启动会议的效果,落实项目启动的质量控制
II范围适用于我院开展的所有申办方发起的临床试验
III规程
1各专业组需至少提前一周邮件发送至机构办邮箱shiyigcp@递交项目启动申请,同时附授权分工表(可参见机构网页样表)、启动会参会人员名单及启动会日程(可参见附件1)
2所有授权研究团队人员必须都参加启动会培训,特殊情况需本人向机构办主任请假
3项目组需根据方案及CFDA相关临床试验的指导原则及法规要求(如:现场核查要点),在启动前准备好所有工作表单后方可申请启动
4授权的研究团队必须具备相应的资质(本院执业医师证)及GCP培训证书,第三方CRC授权必须已签署协议并完成机构办备案
5机构办在接到专业组项目启动申请后必须一周内回复,明确启动会议召开的场地及具体时间
6机构办人员必须参加启动会,熟悉了解临床试验方案和实施要求,并完成对项目启动会的质控,填写《临床试验启动会记录表》(附件2)
7启动会议结束后,专业组/CRA需保留好启动会培训记录、会议纪要、培训材料、启动会签到表、完成的授权分工表等资料
IV参考依据现行GCP第十一章“质量保证”
V附件
1启动会日程
2临床试验启动会记录表
启动会议议程
临床试验启动会记录表(机构)
机构参会人员
年月日。
临床试验管理规程

XXXXXXXXX有限公司一、目的:为了保证新药临床试验过程中遵循科学和伦理道德的原则,使数据的采集、录入和报告做到及时、完整、准确和一致,使受试者的权益和健康得到保护,并保障其安全,保证临床试验遵循己批准的方案、药物临床试验质量管理规范(GCP)和有关法规,使试验结论科学、可靠,根据《中华人民共和国药品管理法》、《药物临床试验质量管理规范》、《药品注册管理办法》、《赫尔辛基宣言》及ICH《人体生物医学研究国际道德指南》等相关法规文件精神,制定本规程。
二、范围:本规程适用于申办者对临床试验过程的管理。
三、责任:研发部。
四、内容:1.临床试验前的准备1.1申办者对临床试验中心的遴选1.1.1申办者在上报药物的临床前研究资料后,根据所申请药物的性质、作用特点、功能主治以及疾病的流行病学、样本量的大小和药品临床试验基地的专业特长等,初步遴选临床试验参加单位和确定参加单位的数量。
1.1.2对初选单位的专业特长、研究资质、人员组成结构、任职行医资格、相关临床试验检查和检测设备以及参研人员参加GCP培训等情况进行现场考察,确认其资质、资源、能力和承担任务量的大小。
1.1.3根据现场考查结果,首先确定临床试验组长单位,经与之协商确立临床试验参加单位,并据此草拟临床试验的《多中心临床试验协调委员会联络表》和《临床试验参加单位初选报告》。
1.1.4国家食品药品监督管理局临床试验批文下达后,申办者根据批文精神,与临床试验组长单位一道最终确定临床试验参加单位。
1.2临床试验文件的起草1.2.1申办者与研究者共同商定起草并签署试验方案、CRF和知情同意书等临床试验文件。
1.2.2申办者起草《研究者手册》,或其替代文件《供临床医师参考的临床前研究药效学、毒理学试验综述》。
1.2.3申办者成立数据和安全监查委员会/数据监查委员会和试验项目小组,根据试验方案设计要求和项目标准由项目小组成员共同制定病例报告表(CRF),监查员可参与部分设计工作。
临床试验方案启动和培训会议记录
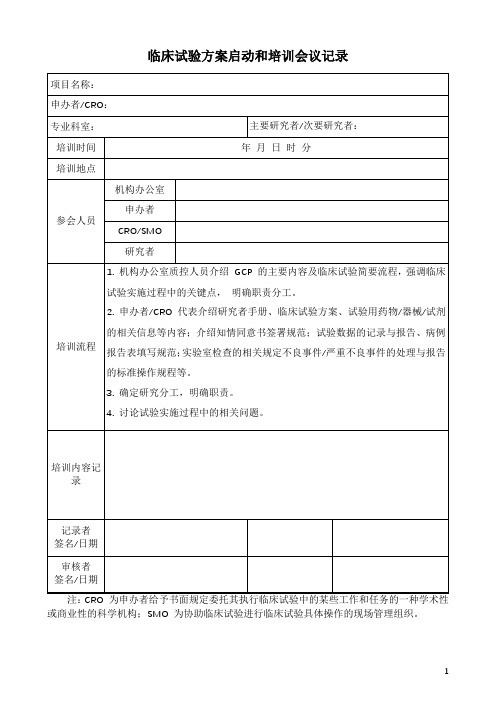
4.讨论试验实施过程中的相关问题。
培训内容记录
记录者
签名/日期
审核者
签名/日期
注:CRO为申办者给予书面规定委托其执行临床试验中的某些工作和任务的一种学术性或商业性的科学机构;SMO为协助临床试验进行临床试验具体操作的现场管理组织。
临床试验方案启动和培训会议记录
项目名称:
申办者/CRO:
专业科室:
主要研究者/次要研究者:
培训时间
年月日时分
培训地点
参会人员
机构办公室
申办者
CRO/SMO
研究者
培训流程
1.机构办公室质控人员介绍GCP的主要内容及临床试验简要流程,强调临床试验实施过程中的关键点,明确职责分工。
2.申办者/CRO代表介绍研究者手册、临床试验方案、试验用药物/器械/试剂的相关信息等内容;介绍知情同意书签署规范;试验数据的记Байду номын сангаас与报告、病例报告表填写规范;实验室检查的相关规定不良事件/严重不良事件的处理与报告的标准操作规程等。
临床试验启动会议纪要

临床试验启动会议纪要Initiation Meeting Minutes Subject:临床试验启动会主题Date:日期Venue:地点Participants:见会议签到表Meeting Attendance Record 出席人员Preparedby:记录员启动会主要内容记录(请根据试验项目要求进行记录,可附页)附件:1、会议签到表2、方案签字页(研究者签名样张)3、任务授权表会议签到表Meeting Attendance Record方案签字页(签名样张)I have thoroughly read and reviewed the study protocol. Having read and understood the requirements and conditions of the study protocol. I agree to conduct or instruct this project and follow the time schedule indicated in protocol. I understand that it is protocol violation if revise the protocol without approval from Ethic committee.我已完全阅读了研究方案,并明白了方案的要求。
我同意遵循方案及时间规程来执导该项临床研究。
我亦知道没有伦理委员会的批准就修改方案是违反方案的。
I agree to perform the clinical study according to the Chinese GoodClinical Practice principles and regulatory authority requirements for source document verification and auditing/inspection of the study.我同意按中国GCP原则进行临床试验,并接受法规部门对原始资料的核查和对临床试验的稽查/视察。
临床试验标准操作规程
第一部分总则第一条:为了保证新药临床试验过程中遵循科学和伦理道德的原则,使数据的采集、录入和报告做到及时、完整、准确和一致,使受试者的权益和健康得到保护,并保障其安全,保证临床试验遵循己批准的方案、药物临床试验质量管理规范(GCP)和有关法规,使试验结论科学、可靠,根据《中华人民共和国药品管理法》、《药物临床试验质量管理规范》、《药品注册管理办法》、《赫尔辛基宣言》及ICH《人体生物医学研究国际道德指南》等相关法规文件精神,制定本标准操作程序。
第二条:药品临床试验依其流程、内容和进程不同,将其划分为临床试验前的准备、启动临床试验、临床试验过程、中期协调会和结束临床试验等五个阶段。
第三条:本标准操作规程是根据药品Ⅱ期临床试验设计要求确立,临床进行的Ⅲ、Ⅳ期临床试验包括部分生物等效性试验均参照本程序执行。
第二部分临床试验前的准备第四条:申办者对临床试验中心的遴选。
⑴申办者在上报药物的临床前研究资料后,根据所申请药物的性质、作用特点、功能主治以及疾病的流行病学、样本量的大小和药品临床试验基地的专业特长等,初步遴选临床试验参加单位和确定参加单位的数量。
⑵对初选单位的专业特长、研究资质、人员组成结构、任职行医资格、相关临床试验检查和检测设备以及参研人员参加GCP培训等情况进行现场考察,确认其资质、资源、能力和承担任务量的大小。
⑶根据现场考查结果,首先确定临床试验组长单位,经与之协商确立临床试验参加单位,并据此草拟临床试验的《多中心临床试验协调委员会联络表》和《临床试验参加单位初选报告》。
⑷国家食品药品监督管理局临床试验批文下达后,申办者根据批文精神,与临床试验组长单位一道最终确定临床试验参加单位。
第五条:申办者起草临床试验文件。
⑴申办者与研究者共同商定起草并签署试验方案、CRF和知情同意书等临床试验文件。
⑵申办者起草《研究者手册》,或其替代文件《供临床医师参考的临床前研究药效学、毒理学试验综述》。
⑶申办者起草《药品临床试验标准操作规程(SOPs)》、《药品临床试验监查工作标准操作规程(SOPs)》、《药品临床试验研究者履历表》、《药品临床试验筹备会议签到表》、《药品临床试验药品发放、回收、清点登记表》、《受试者用药记录卡》和《药品临床试验实验室检查参考正常值范围表》等文件。
药物临床试验之研究者会议
研究者会议
精品文档
在中国GCP中第六十六条中明确规定了:
研“究对于者多会中心议试验组织实施要在临床试验
开始前及进行的中期应组织研究者会 议……”,可见研究者会议的召开也是法 规的要求。
研究者会议Investigator meeting(常用缩写 IM),或“方案讨论会”,主要是各参研 各单位一起共同召开的就研究方案、CRF、 知情同意书和试验操作流程及注意事项等
关键内容达成一致意见的会议,通常由申
精品文档
目的
对方案和临床试验操作流程进行讨论 确保在试验过程中的可操作性 对可能出现的试验问题进行预估和防范 整个项目团队首次见面,彼此熟悉的过程 对整个项目团队进研容床究专背业景领域知识
方案介绍 研究药物介绍 安全性管理 数据管理 中心实验室操作管理 GCP培训 其他:如相关的检查精品技文档 术操作等
文件处理归档
会议日程 签到表 会议纪要 会议材料/PPT Q&A
精品文档
总之,研究者会议我们除开要完善会场布置之外,尽 量把工作重点往方案敲定、培训操作和讨论、统一思 想和项目动员这几个方面靠拢,如果做到了,我们研 究者会议就基本达到了。
精品文档
谢
谢
精品文档
国家药品监督管理局关于调整药物临床试验审评审批程序的公告
国家药品监督管理局关于调整药物临床试验审评审批程序的公告⽂号:国家药品监督管理局公告2018年第50号颁布⽇期:2018-07-24执⾏⽇期:2018-07-24时效性:现⾏有效效⼒级别:部门规章为⿎励创新,加快新药创制,满⾜公众⽤药需求,落实申请⼈研发主体责任,依据中共中央办公厅、国务院办公厅《关于深化审评审批制度改⾰⿎励药品医疗器械创新的意见》(厅字〔2017〕42号),对药物临床试验审评审批的有关事项作出调整:在我国申报药物临床试验的,⾃申请受理并缴费之⽇起60⽇内,申请⼈未收到国家⾷品药品监督管理总局药品审评中⼼(以下简称药审中⼼)否定或质疑意见的,可按照提交的⽅案开展药物临床试验。
现就具体事宜公告如下:⼀、沟通交流会议的准备与申请(⼀)申请⼈在提出新药⾸次药物临床试验申请之前,应向药审中⼼提出沟通交流会议申请,并在确保受试者安全的基础上,确定临床试验申请资料的完整性、实施临床试验的可⾏性。
(⼆)申请⼈准备的沟通交流会议资料应包括临床试验⽅案或草案、对已有的药学和⾮临床研究数据及其他研究数据的完整总结资料。
申请⼈应⾃⾏评估现有的研究是否符合申报拟实施临床试验的基本条件,并明确拟与药审中⼼讨论的问题。
(三)申请⼈应按照《药物研发与技术审评沟通管理办法(试⾏)》(以下简称《沟通交流办法》)要求,提交沟通交流会议申请表(附件1)。
药审中⼼应及时通知申请⼈是否召开沟通交流会议,并与申请⼈商议会议时间。
申请⼈应按沟通交流相关要求按时提交完整的沟通交流会议资料(附件2)。
药审中⼼对沟通交流会议资料进⾏初步审评,在沟通交流会议召开⾄少2⽇前,通过“申请⼈之窗”将初步审评意见和对申请⼈所提出问题的解答意见告知申请⼈。
申请⼈在收到初步审评意见和解答意见后,应尽快反馈问题是否已经得到解决。
申请⼈认为问题已经解决不需要召开沟通交流会议的,应通过药审中⼼⽹站“申请⼈之窗”告知药审中⼼取消沟通交流会议申请;申请⼈认为申请沟通交流的问题仍未得到解决的,按原定计划继续组织会议召开。
临床试验项目启动会准备和召开注意事项
临床试验项目启动会准备和召开注意事项一、启动会前的准备1.对于药物临床试验,申办者应于药物临床试验信息平台登记,并将登记号发送至。
2.对于医疗器械和体外诊断试剂临床试验,申办者应向其所在地省级食品药品监督管理部门备案,并将临床试验备案表(公司盖章)递交至机构办公室。
3.如试验药物由临床试验药房管理,启动会召开前,应向临床试验药房提交药品管理资料及记录。
4.已完成启动前质控。
5.已完成主协议和CRC协议签署。
6.CRC人员已确定,且完成考核和岗前培训。
7.启动会召开前,原则上已完成临床试验方案中所涉及的检验检查项目在GCP系统中的配置。
8.启动会召开前,原则上首付款已到位。
CRA将打款凭证和临床试验经费打款明细表(需为机构表格)发送至,同时妥善安排好物资。
9.CRA与主要研究者协商确认启动会时间和地点。
10.启动会时间和地点确认后,研究者应做好启动会出席人员的通知工作。
出席人员主要为:➢项目研究团队成员,包括主要研究者、研究者、护士、试验产品管理员、CRC等➢申办者/CRO代表➢机构和伦理工作人员➢专业组代表,包括专业组质控员等➢试验过程中涉及的相关辅助科室代表,包括检验科、药学部、病理科等相关科室代表二、启动会的召开启动会议程:➢到会人员签到➢主要研究者或其指定的研究者对试验相关人员进行试验方案相关内容培训➢项目相关的各类细节培训,如试验用药品/器械/体外诊断试剂的管理、样本管理、原始数据记录等➢GCP相关知识培训(如需要)➢提问与讨论➢授权分工与签名留样➢。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
临床试验启动会议纪要
Initiation Meeting Minutes
Subject:
临床试验启动会
主题
Date:
日期
Venue:
地点
Participants:
见会议签到表Meeting Attendance Record 出席人员
Prepared by:
记录员
启动会主要内容记录
(请根据试验项目要求进行记录,可附页)
附件:1、会议签到表
2、方案签字页(研究者签名样张)
3、任务授权表
会议签到表Meeting Attendance Record
方案签字页(签名样张)
I have thoroughly read and reviewed the study protocol. Having read and understood the requirements and conditions of the study protocol. I agree to conduct or instruct this project and follow the time schedule indicated in protocol. I understand that it is protocol violation if revise the protocol without approval from Ethic committee. 我已完全阅读了研究方案,并明白了方案的要求。
我同意遵循方案及时间规程来执导该项临床研究。
我亦知道没有伦理委员会的批准就修改方案是违反方案的。
I agree to perform the clinical study according to the Chinese Good Clinical Practice principles and regulatory authority requirements for source document verification and auditing/inspection of the study.
我同意按中国GCP原则进行临床试验,并接受法规部门对原始资料的核查和对临床试验的稽查/视察。
I agree to use the study material, including medication, only as specified in the protocol.
我同意只应用方案所规定的试验用品包括研究药物。
Signature of Investigator and Study Group 研究小组签字
姓名正楷Printed Name 日期
Date
签名
Signature
任务分配及授权表
Study Center Task Authorization Form
主要研究者签名
/ / Investigator’s Signature:
日期Date:。