3第3章_分离过程中的动力学
第3章 固定化酶催化反应过程动力学

此时,对此微分方程需要根据不同酶动力学特征进行求解。 当酶反应动力学方程为一级反应动力学时, rS =
r ) R ,其中φ= R 3 r sinh(3φ )
rmax CS ,可解得: Km
CS = CS 0
R sinh(3φ
rmax 。 Km iD
当酶反应动力学方程为零级反应动力学时, rS = rmax ,可解得:
φ1
,其中φ1=L
rmax Km iD
当酶反应动力学方程为零级反应动力学时,可解得:
对于球形固定化酶,η0=1 − (
Rc 3 ) , R L 对于膜片状固定化酶,η0 = 1 − c 。 L
当酶反应动力学方程符合 M-M 方程时,无解析解,仅有数值解。 15、梯勒模数φ(Thiele modulus)是表示固定化酶内扩散影响的重要无因次模 型参数,其物理意义为 φ= 表面浓度下的反应速率 。φ值越大,表明内扩散速率 内扩散速率
游离酶 空间效应 本征速率和动力学参数 改变的本征速率和动力学参数 分配效应 固有速率和动力学参数 扩散效应 宏观速率和动力学参数 固定化酶
4、固定化酶催化反应外扩散效应。固定化酶与溶液中底物反应过程包括三步: (1)底物从液相主体扩散到固定化酶表面; (2)底物在固定化酶表面进行反应; (3)产物从固定化酶表面扩散到液相主体。其中酶催化反应速率可由 M-M 方 程 表 示 RSi =
CSi Km r ,= K ,定义Da= max CS 0 CS 0 k L aCS 0
2 CS ⇒ CS +(K+Da-1) CS − K=0 K + CS
1 − CS=Da 可得:
−a ± a 2 + 4 K 解一元二次方程可得: CS= ,其中a = K+Da-1 2 当 a>0 时,取“+”号,当 a<0 时,取“-”号。 (2)有效因子η E 求解。 由定义可知:ηE = 因此, RSi = RS 0ηE
催化作用导论第三章多相催化反应动力学

而不能写成:
பைடு நூலகம்
C、反应机理与反应历程: 反应机理:包括吸附、表面反应、脱附等步骤的序 列称反应机理。 如气-固催化反应机理:
(1)反应物分子在催化剂内表面上吸附;
( 2 )吸附的反应物分子在催化剂表面上相互作用 或与气相分子作用进行化学反应;
(3)反应产物向催化剂内表面脱附。
所要回答的是,反应机理是吸附控制,表面反应 控制,还是脱附控制?
④ 对含活性组分量不同的催化剂样品进行 TOF值的测量, 可以用来作为判别在速率测量中是否存在如传质和 / 或 传热等影响因素的依据; ⑤ 在相同条件下,对暴露不同晶面或有不同晶粒大小的 催化剂样品的TOF值进行测量,可以用于判别晶体各向 异性的重要性。这一点不论在理论上还是在实际上都是 很重要的信息; ⑥ TOF值对开发潜在的催化剂新材料是非常有用的。
1 dn TOF S dt
n = ξ∙NA=TON,S — 活性位数。
在实际应用中,常常用单位活性位的时间得率 STY (site time yield)来表示催化反应的速率。该表示法要 求我们除了要测量催化反应速率外,还要求测量催化剂 的分子数或固体催化剂表面上的活性位数目。优势: ① 如果测量催化反应速率的方法和条件以及测量催化剂 活性位的方法有非常充分的描述,那么不同实验室获得 的同一催化剂的TOF值是完全可以重复的; ② 它也能够用来比较在不同催化剂上获得的 TOF值,例 如,同一种金属的不同形式单晶、金属、负载金属,不 同的金属和不同催化材料的催化剂,从理论和反应机理 研究的意义上讲,这样的比较更具有决定性意义; ③ 即便由于活性位数目测量值的较大误差所得到的 TOF 值只是一个近似值,也能马上判断出该催化剂是不是一 个真正的催化剂。如果 TOF值大于 1则是,如果TOF值 等于或小于 1,则仅仅是一个反应试剂,而催化剂能转 化反应物分子的总数目则是对催化剂的潜在寿命的直接 测量;
第3章水文地球化学作用过程中地反应动力学

.第三章 水文地球化学作用过程中的反响动力学在水文地球化学作用的研究中仅有化学平衡的概念是不够的,在许多情况下还需要有反响动力学的概念.本节介绍有关反响动力学的概念和原理,说明为必须要考虑反响动力学,并讨论在情况下必须将化学平衡学和反响动力学两种概念结合起来考虑.加深对于水-岩体系中的平衡和动力学概念的了解,对提高我们对地下水的化学成分和同位素组成的理解是十分有益的.只有掌握知识才能更好地管理好水资源,更好地解决生产工作中的各种与地球化学有关的问题.化学平衡的原理可以用来确定水文地球化学作用的方向、程度和边界条件,水溶液中某种化学成分的平衡浓度.而化学动力学的原理可以用来确定该水文地球化学反响达到平衡状态所需要的时间与其反响途径,反响所处位置,各反响途径所需的时间.只有当地下水在水-岩体系中所停留的时间大于水文地球化学作用达到平衡状态时所需要的时间,化学平衡的概念足以用来理解和确定该体系的水化学性质,例如,深层承压水盆地可归属这类体系.当水文地球化学作用达到平衡状态时所需要的时间,明显地长于地下水在水-岩体系中所停留的时间,如此仅依靠反响平衡原理来研究地下水的化学性质是不够的,此时需同时应用化学平衡和反响动力学的原理,例如,地下水位很浅的体系就属于此类情况.由于生产和环境工作中的需要,人们注意到必须对地球化学中的反响动力学进展研究,八十年代以来开始做了大量工作.目前在运用化学平衡<包括吸附作用>来解释天然水的化学行为方面已相当有效,但对水-岩体系中的化学动力学的理解相对较为不足,目前在水文地球化学研究中仅应用了最为简单的反响动力学的概念.第一节 化学动力学的某些根本概念一、体系状态根据体系与环境的关系,热力学体系可分为三类:①开放体系:与环境既有物质交换,又有能量交换的体系称为开放体系,如潜水区包气带水、地下水排泄地段、地球化学栅地段.②封闭体系:与环境之间只有能量交换,没有物质交换的体系称为封闭体系,这种体系可以改变自己的容积,如铁轨的热胀冷缩,水的冻胀融缩.热水给围岩热量,或围岩地热对地下水的加温〔在水与围岩之间没有发生物质交换的条件下〕都是封闭体系的现象.③孤立体系:与环境之间既无物质交换也无能量交换和容积变化的体系称为孤立体系.体系的状态可分为平衡状态,稳定状态和图 3.1.1开放型地下水体系中水溶相反响A=B 的状态 dn A —发生在大气、水溶和固相间的A 通量;C A —A 物质的浓度;n a —A 物质的摩尔数;V —水相的体积.〔据D. Langmuir, 1985〕. 非稳定状态.大局部深层承压水和某些深层非承压水的化学成分长期处于稳定不变的状态,这种状态可以认为是热力学平衡状态, 水-岩体系可认为是一种封闭体系,因此,可以安全地应用平衡概念来说明其化学性质.然而在的些浅层承压水和潜水中,其水化学成分可以在没有达到平衡状态条件下处于某种不变化的状态,这种体系在热力学概念上虽是开放的,但是水化学成分处于一种稳定状态.非稳定状态是水化学成分随时间而变化.图是地下水体系中水溶相反响的环境示意图.水溶相反响处于平衡状态、稳定状态和非稳定状态的条件如下1:dn A = dn A ’, dn A ’/dt = 常数封闭体系 平衡状态 dn A ≠ dn A ’, dn A ’/dt =常数开放体系 稳定状态 dn A ≠ dn A ’, dn A ’/dt = 变量 开放体系 非稳定状态二、反响半和水的滞留时间水的停留时间是指水进入体系至由该体系中出来所经历的那段时间.在开放的均一的体系中,水的停留时间的计算公式如下T R =V r式中T R —水的滞留时间,V —混合均一的水的体积,r —水流的速率.假设一个简单的一阶反响,A ↔当达平衡状态时,K AB =k k +- 式中k +,k - 是正向和逆向反响的速率常数.反响的半期是指反响进展到反响物的被消耗为其初始浓度一半所需要的时间.一阶反响的半期的计算公式为,T 1/2=ln .20693k k ++= 式中T 1/2—反响的半期.假设B 的初始浓度=0,如此可写成:C C K T T A B AB R =+1069312.当T R >>T 1/2,式<3.1.5>便变成为平衡表达式,因而在这种条件下,平衡概念可被采用.与此相反,当T R ≤T 1/2,必须采用反响动力学概念方能对一个反响的状态作出解释. 大局部地下水的化学反响的半期处于10-10~ 106秒,也有以年为计,或更长.所以,热力学和反响动力学对研究水-气体系的化学成分和同位素来说都是极需要的. 图表示了不同水体的滞留时间和某些反响的反响速率<度>之间的关系.表 3.1列出了某些反响的半期的例子.图水圈中某些水体的半反响期<T 1/2>和滞留时间<T R ><据D. Langmuir, 1985>表某些反响的例子和它们的近似反响半周期 <据D. Langmuir, 1985>反响类型 半反响近似值 1 水合. H 2O+CO 2<aq>=H 2CO 3 K eq =[H 2CO 3]/[CO 2] ~1秒 2酸碱反响〔包括离解反响〕H 2CO 3 = H + HCO 3 K eq = [H +][HCO -3]/[H 2CO 3] ~IO -6sec.HCO 3- = H + + CO 2-3 K eq = [H +][CO 2-3] 10-6 sec. 3络合反响Cu 2+ + H 2O = CuOH + + H + ~10-10 sec.Fe<H 2O>62+ = Fe<H 2O>52+ + H 2O ~10-7 sec. 4聚合和水解Al<OH>n 3-n +<3-n>H 2O → Al<OH>3+<3-n>H + 数月 5放射性衰变14C →14N +e - 5,570年 6同位素交换34SO 2-4+H 32S 2—→H 34S -+32SO 2-4 数月—数年 7气体溶解和逸出CO 2<g>=CO 2<aq> K eq =[CO 2]/P CO2 T 正向<T 反向, 数秒—数小时 8氧化复原Fe 2++1/4O 2<g>+5/2H 2O=Fe<OH>3+2H +Keq = [H +]2 /[Fe 2+][P O2]1/4 数分—数小时9 吸附—解吸Cd 2++CaX = Ca 2++CdX ][][][][22CaX CdX Cd Ca K ex ⋅=++ 数秒10 沉淀—溶解Ca 2++HCO - = CaCO 3 + H + Keq = [H +] / [Ca 2+][HCO -3] 大于数周11 矿物结晶<针铁矿>Fe<OH>3·nH 2O<隐晶状>→α-FeOOH 〗<针铁矿>+<n+1>H 2O数年 图说明在雨水中很少反响能达到平衡状态,温度和压力仅在几秒中或更短的时间内处于不变状态.在雨水中可能处于平衡状态的只有某些溶质-溶质或溶质-水之间的反响,如酸-碱反响和离子化学反响.雨水与小颗粒之间的吸附反响可能处于平衡状态.吸附作用的T 1/2线右边是虚线箭头,指示T 1/2可达几天.这种时间较长的吸附作用是由于它包括有进入岩石碎屑中的扩散作用,所以扩散作用的T 1/2控制了吸附平衡的实现.地下水的滞留时间的主要X 围从岩溶型碳酸盐和具高渗透性的浅层砾石层中的几天到深埋的承压水盆地中的几个百万年.显然可以设想大局部在深部沉积盆地地下水中的化学反响是处于平衡状态的.放射性衰变率<一般是不可逆的>的时间X 围由不到1秒至数十亿年.同位素交换的速率的分布X 围也相当宽广.三、反响动力学概念反响动力学在水-岩体系中的应用不如化学平衡原理应用得好,而且应用反响动力学原理并不那样容易,有些作用受矿物颗粒的外表特征,矿物外表上被吸附的痕量物质和有机物活动的影响很大<Berner 1978>.由于在实验室中配制的矿物颗粒的活性比天然状态强,所以实验室中矿物溶解的速率要快几个数量级;天然水中不溶的<Salution in hibit>微量物种<如磷酸盐>的吸附作用也是很快的,实验室中的氧化复原反响,例如Fe<Ⅱ>要比自然状态慢许多,因为自然界中有微生物的催化作用.但是人们还是很关心解决水岩作用过程中的化学动力学问题.Claassen 1981,Paces 1983曾对地下水体系中矿物/沉淀/溶解的反响动力学原理进展过研究.以下对反响类型、速率定律、速率常数与温度的关系与绝对速率等问题予讨论.1.反响类型在动力学中单元反响和全反响是有原如此性差异的.反响物、生成物在分子水平上发生的反响称为单元反响,单元反响是不能再分解的反响.全反响如此是由两个以上的单元反响组成的.例如,H++OH-= H2OCO2<aq>+OH-=HCO-3H4SiO4=SiO2<石英>+2H2O它们都是单元反响<elementary reactvon>,单元反响的反响物和生成物可以是离子,分子,离子团或自由原子.臭氧的分解是全反响,2O3 3O2k+1k+2它包括二个单元反响, O3 O2+O与 O+O3 2O 2k-1k-2式中中间形式之一是自由氧原子<Lasaga 1981>.人们经常假定中间反响具有很灵活的过渡形式,在全过程中,它们形成和消失的速率很快变为相等.这对臭氧分裂也是正确的,对k-2<<k+2和稳定状态条件的反响是正确的.d<O>/dt = k+1<O3>-k-1<O2><O>-k+2<O><O3> = 0稳定状态的假设可以帮助和求解氧原子的浓度,因而可以在下一步的速率公式计算将它排除<Lasaga 1981>.另一个有关假定是当全反响<overall reaction>处于平衡状态时,如此该全反响中的正向单元反响和逆向单元反响的速率必定相等,这一原如此称为微观可逆性.单元反响的途径可由反响式清楚地知道,但全反响的途径如此不然.全反响的速率是由它的组成局部单元反响的速率所决定的,只有组成它的单元反响的速率知道后,全反响的速率才能被确定.单元反响的速度总是与其反响物的浓度成正比,而全反响的速度如此不然,全反响的速度一般是由决定性反响阶段的速率所控制的.全反响有两种:串联反响和并联〔平行〕反响.对串联全反响<Sequential reaction>,如HCO-3+H+=H2O+CO2<aq>CO2<aq>=CO2<g>第一个反响十分快,不到一秒钟就已完成,而第二个反响要慢几个数量级,所以后者是速率决定性的反响.对平行反响来说,最快的反响是速率决定性反响.例如,黄铁矿被溶解氧的氧化相对较慢,FeS2<s>+7/2O2<aq>+H2O=Fe2++2SO2-4+2H+但被Fe3+离子的氧化速率相对快,FeS2<s>+14Fe3++8H2O=15Fe2++2SO2-4+16H+因此,第二个反响式是速率决定性的反响.由于速率决定于浓度,所以速率决定性反响可以随浓度的变化而变化.又如,在酸性水中碳酸钙的溶解速率从属于pH值,但在高pH值介质中它的溶解.速率从属于H2CO3的浓度.<Plummer 1979>2.速率定律:对理想单元反响A=B,其正向反响A→B的速率R+= dA/dt = k+<A>,逆反响B→A的速率R- = dB/dt = k-<B>,如此单元反响的速率在总体上为,正向速率R+ = k+∏ <A i>n<A—反响物逆向速率: R-=k-∏ <B i>n<B—生成物当反响平衡时,k+/k-=∏ <A i>n eq=K eq, <A—反响物和生成物单元反响速率的阶数是由参于反响的原子和分子的当量数所决定的,例如反响,A+2B=C反响速率-dC/dt = k+<A><B>2,所以该反响速率是三阶.某些对地球化学有关的反响速率的经验公式列于表.表总结了一些速率公式的积分和微分形式,零阶反响的半公式t1/2=0.5A0/k+一阶反响的半公式为: t1/2=0.695A0/k +表某些简单阶数反响的速率公式与其积分形式〔据: D. .Langmuir, 1985>零阶反响:A0<aq>→A<g或s>-dA/dt = k- dA/dt = k +A = A0+k-t A = A0-k+t一阶反响:A<aq>→B<aq>-dA/dt = k+AdA/dt = k+<A s-A> lnA = lnA0-k+tln<A s-A> / <A s-A 0> = -k+t 二阶反响:A+B→C<1/v A><dA/dt> = k+A2 <V A= -1或 -2>-dA/dt = k+<A><B>1/A=1/A0-V A k+tln[<A0><B>/<B0><A>] = <B0-A0>k+t注:上标‘o’表示时间为零时的初始状态;下标‘s’表示反响达到平衡时的饱和状态3.速率常数从属于温度速率常数与活化能有关.Arrhenuis根据实验测量的结果得出:k=Aexp<-E a 式中A—称为A因子,A与T稍微有关,可以忽略T的影响.E a—活化能,它总是正值,这说明随T增高,反响速率也相应增高.dlogk/dt = E a/2.30RT著名的大姆指法如此是在温度为25℃左右时,每<增高>10°C,速率翻倍与此相应在25°C时活化能为12kcal/mol.对公式取对数:lnk = lnA-E a/RT或logk = logA-E a/2.30RT〔〕..一般评估Ea 的方法是作logK 与1/T 的座标图,图,是硅酸盐岩和矿物的溶解—沉淀速率图和石英、隐晶SiO 2的沉淀图2.图石英和隐晶二氧化硅的logk 对1/T 图 <据Rimstidt 和Barnes, 1980>活化能有益于理解矿物溶解和沉淀反响机理的实质,水扩散控制的反响活化能<5~6kcal/mol<Berner 1978,Rimstidt1981>,由外表反响控制的矿物溶解—沉淀作用的E a 一般为10~20kcal/mol<Lasaga 1981>.矿物固体态扩散的E a 在低温条件下为20~120kcal/mol,一般在20~80kcal/mol 之间.Apps1983测得透辉石<obsidin>和武岩玻璃由扩散作用形成的蚀变作用具有E a ≈22—130kcal/mol,物理吸附作用的E a =2—6kcal/mol,但化学吸附的Ea 一般都大20kcal/sags 1981年指出,因断键或生成键所引起的外表反响而控制的矿物溶解和沉淀作用,可以预料它们的活化能与固相扩散作用相仿.吸附作用的活化能是如此低,故化学吸附—解析作用将比矿物的溶解和沉淀作用优先进展.换句话说,化学吸附热将降低外表作用控制的矿物作用所需要的E a .4. 绝对速率<过渡态>理论和活化的络合物深入思考反响机理得出绝对速率理论的概念.这一理论基于二个假定:<1>在一个反响中的反响物与生成物之间存在着一个最大能量垒,而活化的络合物存在于这最高能量垒上〔图〕;<2>在反响物生成物和活化的络合物 <C ±>之间存在有一定的化学平衡状态,反响速率决定性阶段相应于活化的络合物的形成和瓦解, k + A+B ↔C ±↔AB k R = <k B T/h>K ±[A][B] k o ’ = <k B T/h>K ±式中k B ,h 为Boltzman 和Planck 常数; k o ’为经活度校正的绝对速率.k’是没有进展离子力校正的反响速率常数. ))(()( ''B A o k k γγγ±⋅= 应用活度系数计算公式可以离子力对二阶反响速率常数的影响,简化或得,假设A 、B 其一样的价态符号,如此k'随I 增高而增高,图表示,当I=6×10-3m 时,包括二价离子A 和B 二价反响的速率,将是纯水中的两倍.图3.1.3 各种硅酸盐岩石和矿物的logk 对1/T 图 <据D. Langmuir, 1985>图3.1.5 在A+B=C ±时,AB 反响中被活化的络合物的自由能〔据D. Langmuir, 1985〕 图3.1.6 在反响物A 和B 的不同价态的乘积时<即Z A ×Z B >,离子力对二阶速率常数影响的预计<在I=0时,k’=k o ’ 据D. Langmuir 1985〕.第二节SiO 2沉淀作用的反响动力学计算实例运用化学动力学原理解释水中高浓度超饱和SiO 2含量在两年内没有发生沉淀的原因.水溶相化合物之间的反响速率是较快的,因此很容易达到平衡状态.水溶相与固相之间的反响<溶解和沉淀作用>和氧化-复原作用的反响速率较慢,有时甚至需要很长时间才能达到平衡状态.水中SiO 2的沉淀作用便是个典型例子.例如,有个水样中SiO 2浓度达150mg/l,水样的pH=3.水中SiO 2与结晶石英之间的平衡含量1.2mg/l,与隐晶质SiO 2之间的平衡含量较高,为116mg/l,低于水中SiO 2浓度,即该水中SiO 2样浓度处于超饱和状态.但是水样中SiO 2浓度在室温25℃条件下保持了两年没有发生变化.在分析测试等方面无过失的情况下,其原因只能从化学动力学方面予以探索.SiO 2溶于水的反响方程为:SiO 2<am>+2H 2O=H 4SiO o 4logk +=-0.369-7.89×10-4T -3438/Tlogk -=0.707-2598/T当t=25℃时,k +=10-12.14sec -1 , k-=10-9.42sec -1<Morey,Foutnier 和Rowe's,1964>. 按Langmuir 和Mahoney<1985>关于SiO 2<an>的溶解〞沉淀动力学计算公式: 式中:A -暴露于溶液中固相的相对外表积,m 2;M -水的质量,kg.假设A/M=1,如此按上式可算得: d H SiO dt m o [](()(.))./sec ..4412199423131010251022610=-⨯=-⨯----·L 假设A/M=100,如此,以上计算所得仅仅是SiO 2的初始沉淀速度.Rimstidt 和Barnes 于1980年提出了计算SiO 2沉淀时间的公式.t k k k H SiO k k H SiO =----+-+-14404400ln[[][]] [H 4SiO 04]是时间等于t 时的浓度,而[H 4SiO 04]0是时间为零时的浓度,如此t A M k k k H SiO k k H SiO o (sec)ln[[][]]=-⎛⎝ ⎫⎭⎪⎪⎪---+-+-1440440 根据上式计算在温度=25℃时,[H 4SiO 04]浓度从150mg/L 变化至115mg/L 所需要的时间.当A/m=1时, t=1.28×1010 t=407yr而当A/m=100时, t=1.28×108 t=4.07yr以上计算可以用来解释为上述水样中SiO 2的浓度保持了两年而没有发生明显的SiO 2沉淀作用.小 结地球化学作用可以改变岩石的性质,也可使地下水的变得更有用或不适用.对化学平衡和动力学的理解可大大提高人们对地下水的化学成分和同位素成分的能力,因而更好地管理水资源.因此,水文地质工作者需要对地下水的地球化学有所了解.化学平衡决定了反响条件的边界,例如,某个存在形式的最高或最低浓度是多少?化学动力可用来决定一个反响需要多长时间才能达到平衡状态,与通过哪些反响途径达到平衡状态的.只有当反响速率快于地下水停留时间时,单一平衡原理才能有效,它对深层承压水的研究最有应用价值,当反响速率与地下水停留时间相当或小于它时,如此需要同时应用平衡学和动力学两套原理,这对水位浅的水-岩体系特别重要,水化平衡概念<包括吸附>,已成功地应用于假释地下水体系的许多化学现象,但对动力学的理解相对不足,目前只有简单的动力学概念在地下水研究中得以成功地应用.反响速率常数受温度和矿化度〔离子力〕的影响.复习思考题1.为在水文地球化学中要研究反响动力学?2.在条件下必须采用反响动力学的原如此?3.平衡状态与稳定状态有何不同?4.表示各种天然水在体系中的滞留时间.5.利用图2和表1说明那些反响的速率较快,那些反响的速率慢.6.叫单元反响,是全反响?7.决定全反响的速率是那种单元反响?8.反响速率的阶数是根据确定的?14C→14N + e的反响阶数?9.写出零阶和一阶反响的浓度与时间的关系式.10.说明反响速率与温度的关系.回答为?11.说明反响速率与矿化度的关系.回答为?12.是绝对速率?参考文献1 D. Langmuir,Aqueous Environmental Geochemistry, Prentice Hall Upper Saddle River, NJ07458,p.50~582 ngmuir, J. Mahoney, Chemical Equilibrium and Kinetics of Geochemical Processes in Groind Water Studies, Practical Applications of Ground Water Geochemictry, Proc. First Canadian/American Conf. On Hydrogeology, 1985.。
第三章化学动力学初步

限速步骤(定速步骤):在一连串的反应中,较慢的基元反应限 制 了整个反应的速率,这一步骤称为限速步骤。
4
3.2 反应速率理论简介 速率理论简介 论简
3.2.1有效碰撞理论
1.反应速率正比于反应物分子的碰撞次数υ∝Z0c(NO)c(O3) 2.反应物分子必须定向碰撞才可能发生反应 有效碰撞
无效碰撞
υ∝PZ0c(NO)c(O3)
10
3.4 反应速率与温度的关系
3.4.1 范特霍夫 范特霍夫(Van’t Hoff)近似规则 近似规则 近似
反应体系的温度每升高10 K,化学反应速率将增加到原来的 k T + n ⋅10 2~4倍。 k T + 10 =γn =γ
kT
kT
γ:温度系数;一般为2~4,k:各温度下的反应速率常数。
9
表3.2 cA和cB及反应速率v
∴根据实验结果可知:m = 2,n = 1 反应的速率方程为υ =kcA
2c D
反应的级数:m + n =
3
说明 (1)常见反应级数:零、一、二级反应,三级反应少见,三级以上 反应无。 (2)反应级数可为零、正整数、分数, 反应级数不同K的单位不同。 (3)复合反应可由限速步骤写出速率方程。
单位是J·mol-1 。
上式两边取自然对数,得:
Ea ln k = ln A − RT
Ea lg k = lg A − (3.6) 2.303RT
【例3-1】对下列反应C2H5Cl(g)→C2H4(g) + HCl(g),已知 A=1.6×1014,Ea=246.9 KJ·mol-1,求700 K时的速率常数。如果 反应温度升高到710 K时,计算速率常数,并与700 K时的速率常 数做比较。
环境工程原理知识点总结

环境工程原理知识点总结第II篇思考题第一章绪论1.“环境工程学”的主要研究对象是什么?2. 去除水中的溶解性有机污染物有哪些可能的方法?它们的技术原理是什么?3. 简述土壤污染治理的技术体系。
4. 简述废物资源化的技术体系。
5. 阐述环境净化与污染控制技术原理体系。
6. 一般情况下,污染物处理工程的核心任务是:利用隔离、分离和(或)转化技术原理,通过工程手段(利用各类装置),实现污染物的高效、快速去除。
试根据环境净化与污染防治技术的基本原理,阐述实现污染物高效、快速去除的基本技术路线。
第二章质量衡算与能量衡算第一节常用物理量1.什么是换算因数?英尺和米的换算因素是多少?2.什么是量纲和无量纲准数?单位和量纲的区别是什么?3.质量分数和质量比的区别和关系如何?试举出质量比的应用实例。
4.大气污染控制工程中经常用体积分数表示污染物的浓度,试说明该单位的优点,并阐述与质量浓度的关系。
5.平均速度的涵义是什么?用管道输送水和空气时,较为经济的流速范围为多少?第二节质量衡算1. 进行质量衡算的三个要素是什么?2. 简述稳态系统和非稳态系统的特征。
3. 质量衡算的基本关系是什么?4. 以全部组分为对象进行质量衡算时,衡算方程具有什么特征?5. 对存在一级反应过程的系统进行质量衡算时,物质的转化速率如何表示?第三节能量衡算1.物质的总能量由哪几部分组成?系统内部能量的变化与环境的关系如何?2.什么是封闭系统和开放系统?3.简述热量衡算方程的涵义。
4.对于不对外做功的封闭系统,其内部能量的变化如何表现?5.对于不对外做功的开放系统,系统能量能量变化率可如何表示?第三章流体流动第一节管流系统的衡算方程1.用圆管道输送水,流量增加1倍,若流速不变或管径不变,则管径或流速如何变化?2.当布水孔板的开孔率为30%时,流过布水孔的流速增加多少?3.拓展的伯努利方程表明管路中各种机械能变化和外界能量之间的关系,试简述这种关系,并说明该方程的适用条件。
第三章 化学反应动力学的计算
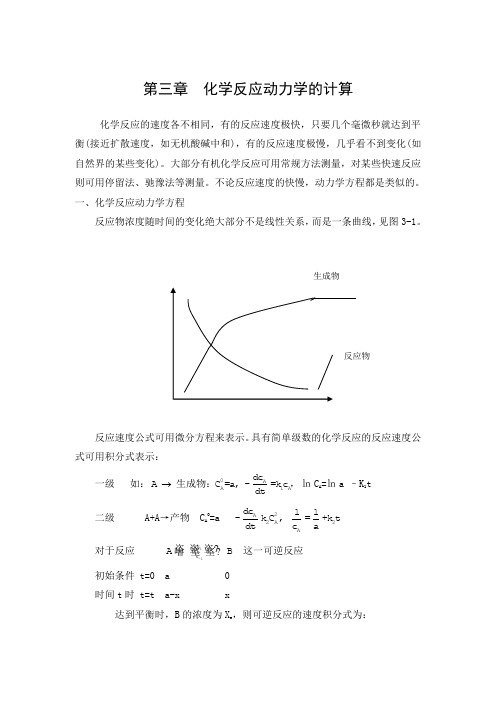
第三章 化学反应动力学的计算化学反应的速度各不相同,有的反应速度极快,只要几个毫微秒就达到平衡(接近扩散速度,如无机酸碱中和),有的反应速度极慢,几乎看不到变化(如自然界的某些变化)。
大部分有机化学反应可用常规方法测量,对某些快速反应则可用停留法、驰豫法等测量。
不论反应速度的快慢,动力学方程都是类似的。
一、化学反应动力学方程反应物浓度随时间的变化绝大部分不是线性关系,而是一条曲线,见图3-1。
反应速度公式可用微分方程来表示。
具有简单级数的化学反应的反应速度公式可用积分式表示:一级 如:0AA1Adc A C =a, -=k c dt 生成物:,㏑C A =㏑a –K 1t 二级 A+A →产物 C A 0=a 2A 2A 2A d c 11-k C , =+k t d t c a对于反应 1-1k k A B 这一可逆反应初始条件 t=0 a 0 时间t 时 t=t a-x x达到平衡时,B 的浓度为X e ,则可逆反应的速度积分式为: 级数:1-1 1-10k A A e e 1A -1B k 0e 0C =a dc x xA B=-k C +k C : =kt dt a x -xC =0ln 1-21-10Ak0A e e e B 1A -1B C k e e 0CC =a dc x ax +x(a-x )A B+C C =0=-k C +k C C : =kt dt 2a-x a(x -x)C =0ln 二、常微分方程的解化学反应动力学方程是用微分方程表示的,对于简单的反应,可直接求得微分方程的解。
微分方程:()(1)(,,,......)......(1)n n y f x y y y -'=在区间a<x<b 的解,是指()y x ϕ=,这样一个函数,在所述区间内存在导数()(),(),......()n x x x ϕϕϕ'''。
且对于区间a<x<b 内的每一个x ,等式(1)都成立。
第三章(3)__液相传质步骤动力学解析

nF
R
c
s R
1.反应产物生成独立相
s R
cRs
s R
1
∴
= 0+ RT
nF
ln
cs
0O
由于:
cOs
1
i id
cO0
∴
= 0+ RT
nF
ln
0 cO0 1
i id
平+
RT nF
ln 1
i id
反应产物生成独立相时的极化曲线
2.当反应产物可溶时
有时,阴极电极反应的产物可溶于电解液,或者生成汞齐Ji CiVx
xl
Ji
Di
dci dx
电极表面传质区域的划分
c
cs
c
双电层区
x0
x1
d
扩散区
c c0 c0 cs
对流区
x2 x
3.3.2 稳态扩散过程
一. 理想条件下的稳态扩散
c c 0
s
Ag
Ag
强烈搅拌
管径极小
dc c0 c s 常数 dx l
K
Ag
NO3
大量局外 电解质
ln oo DR R R Do
常数
(3.15)
由于在一定对流条件下的稳态扩散中,o与R均为常数; 又由于在含有大量局外电解质的电解液和稀汞齐中,
o R Do DR均随浓度co和cR变化很小,也可以将它们看 作常数,因此可以将1/2看作是只与电极反应性质(反 应物与反应产物的特性)有关、而与浓度无关的常数。
通过控制转速,模拟不同 值的扩散控
制的电极过程 。
四.电迁移对稳态扩散的影响
以 AgNO3 溶液为例
i 2FD
第三章 细胞反应动力学

四、胞内代谢反应
根据功能分为: 供能反应 生物合成反应 多聚反应 组装反应 根据过程分为: 初级代谢 次级代谢
五、胞内代谢调控
实质 把细胞内所有酶组织起来,通过活化某些酶、抑 制另一些酶,甚至出现一些新酶,去掉某些原有的酶, 以使整个代谢过程适应细胞生理活动的需要
两个重要机制 酶活性调控 酶合成调控
cS cS max exp( ) K S cS K SI cS cS ) exp( )] Teissier等: max [exp( K SI KS
三、有抑制的细胞反应动力学
产物抑制 对产物竞争性抑制:
max cS
cP cS K S (1 ) K PI
三、有抑制的细胞反应动力学
底物抑制 对底物非竞争性抑制:
d max, 0 dcS
* cS KSI KS
*
max
1 2 K S / K SI
三、有抑制的细胞反应动力学
底物抑制 对底物竞争性抑制:
经验方程 Aiba等:
max cS
cS cS K S (1 ) K SI
cS 为限制性底物的质量浓度,g/L K S 为饱和常数,g/L
二、无抑制的细胞反应动力学
Monod模型方程
cS
二、无抑制的细胞反应动力学
Monod模型方程
不同K S值的Monod曲线
二、无抑制的细胞反应动力学
Monod模型方程 max 和 c S 为一级动力学关系 cS , K S时, 当 cS KS 提高限制性底物浓度可以提高比生长速率
13401370436生物反应工程第三章细胞反应动力学概述研究对象以细胞微生物催化剂的反应过程动力学研究内容在细胞水平上通过对细胞的生长速率代谢产物的生成速率和底物的消耗速率等动力学特性的描述反映出细胞反应过程的本征动力学特性研究目的细胞反应过程动力学是进行细胞反应过程优化和生物反应器设计的重要理论依据主要内容第四节底物消耗和产物生成动力学第一节细胞反应概论一基本概念细胞细胞是一切生物体进行生长遗传和进化等生命活动的基本单位也是决定生物体形态结构和功能的基本单位代谢产物排泄进入胞外非生物相二细胞的基本特征组成chon四种元素约占细胞质量的90spnacakclmgfe含量其次以上12种元素约占细胞质量的99细胞的化学组成二细胞的基本特征组成活细胞的主要成分是水占总量8095干物质中90是由蛋白质核酸糖类和脂类等四类大分子物质所组成细胞的元素和化学组成将直接影响细胞大规模培养时的培养基设计二细胞的基本特征组成蛋白质
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
现代分离方法与技术-第3章:分离过程的动力学
3.2 流体的迁移与扩散
在多数情况下,溶质是在流体(液体、气体、超临界流体)
中迁移或扩散的。如果流体本身不流动的话,溶质在流 体中就会因为浓度梯度而迁移(或扩散)或者因为其他场 的作用定向迁移; 在分离技术中,流体本身也迁移的情况常常碰到(如色谱 分离),而且流体多数情况下是沿管路流动的; 流体的流动性能与其黏度密切相关。流体的黏度是流体 的一部分阻止另一部分移动的阻力,当流体处于流动状 态时,在相同外界条件下,黏度就决定了流体流动的速 度。
现代分离方法与技术-第3章:分离过程的动力学
将式(3.21)代入(3.20)得: (p1 -p2 )(r 2 -r'2 ) = (3.22) 4L 所以,作为r'函数的流速的层状图呈抛物线形。 在时间t内,流过半径为r'和r' + dr'两表面间横截面上的 体积为: dV =2 tr'dr' V = 2 tr'dr'
现代分离方法与技术-第3章:分离过程的动力学
在低浓度下(假定 =c),化学势可以写成
= ext + +RT ln c
式中, ext为外场作用于分子(粒子)上的化学势; 为 体系的标准化学势;c为溶质浓度,假定其在扩散方向 不随时间变化,于是有: c d c d( ext + +RT ln c) J =- =- dx f dx f c d ext d RT dc =- + + dx c dx f dx c d ext d RT dc =- + dx f dx f dx
现代分离方法与技术-第3章:分离过程的动力学
式(3.13)即为普朗克 - 爱因斯坦(Planck - Einstein) 方程,有: RT dc dc J = =-D (3.14) dx f dx 式(3.14)即为费克(Fick)第一扩散定律,是早在1855年就 已发表的描述分子扩散的基本方程。费克第一扩散定律 的通式可以写成: dy J (x)=-A (3.15) dx dy 式中,J(x)表示沿x轴方向的流;A为比例系数; 指在x轴 dx dc 方向上的梯度。当y为浓度c时, 为x轴方向上的浓度梯度。 dx
现代分离方法与技术-第3章:分离过程的动力学
因为分子或离子的运动速度难于测定,所以人们提出了 一个易于测定的物理量-流密度(J )-来反应分子在液体中 的运动速度。流密度是指单位时间内通过单位面积的物 质的量(mol)。 当分子运动的平均速度U的单位为cm/s,溶质的浓度c的 单位取mol/cm3时,有下列关系式: J =Uc c d f dx (3.10) 将式(3.9)代入(3.10)中得: J =(3.11)
现代分离方法与技术-第3章:分离过程的动力学
与宏观物体的机械运动相比,溶质分了在溶液中的运动 加速度几乎为零(几乎是立即达到稳态)。因此,处理分子 运动时可以认为其平均加速度为零,即: d2 x M 2 =0 dt 由分子运动方程可得: d 2 x d dx M 2 =-f =0 dt dx dt 分子运动的平均速度为: d x 1 d U = =- dt f dx (3.9)
现代分离方法与技术-第3章:分离过程的动力学
3.1 分子迁移-费克第一扩散定律
所有溶质的迁移都是朝着趋向平衡的方向进行的,平衡控
制着组分的迁移方向。但仅用平衡的观点无法准确回答组 分的迁移速度和迁移性质的问题,而迁移速度与整个分离 速度是密切相关的; 物质的输运过程是指在适当的介质中,在化学势梯度的驱 动下物质分子发生相对位移的过程; 物质的扩散运动也是在梯度(浓度梯度)驱动下,物质分子 自发输运的过程; 扩散速度的差异可使某些组分达到分离,但也会使组分的 谱带展宽; 无论是定向迁移还是非定向扩散,涉及的都是物质分子的 迁移,因此分子迁移的表征是研究分离过程的基础
现代分离方法与技术-第3章:分离过程的动力学
上式中以1mol物质计,M 为物质的摩尔质量; x表示整个 1mol分子(粒子)运动的位移平均值;表示每摩尔运动着 f 的分子的阻力常数,即摩尔摩擦系数,它是所有分子的 摩擦系数f 的加和,即 f =N A f 。 所有分离过程都在不同程度上对物质(溶质)进行输运, 分离所需时间与输运时间有关,因而也与摩擦系数有关。 Stokes定律描述了分子运动摩擦系数与其它因素的关系, 即:f =6 N A r (3.8) 式中,为摩尔摩擦系数; f N A为阿伏加德罗常数;r为球形 溶质分子的半径;为介质黏度。
(3.25)
现代分离方法与技术-第3章:分离过程的动力学
黏度系数受温度的影响较大,而且气体和液体的黏度 系数受温度影响的程度相差很大。实验表明液体的黏 度系数的关系可以近似表示为:
=Ae E/RT
(3.26)
式中,A为常数;E为流的活化能,即流速取决于流物 质分子通过能垒的特性 气体的黏度系数可表示为:
现代分离方法与技术-第3章:分离过程的动力学
最终通过毛细管的流体的总流量Q可表示为:
r 4 p Q= 8 L
(3.30)
现代分离方法与技术-第3章:分离过程的动力学
3.3 带的迁移-费克第二扩散定律
费克第一定律是假设溶质浓度c在扩散方向上不随时间变
化,但实际上溶质浓度c在扩散方向上是随时间变化的
现代分离方法与技术-第3章:分离过程的动力学
dp F1 =(3.4) dx dx F2 =-f (3.5) dt 将式(3.4)和(3.5)代入(3.3)。得到宏观物体基本运动方程: d 2 x dp dx m 2 =- -f (3.6) dt dx dt 如果将宏观物体运动的方程用来描述分子运动,则得到 分子运动方程为: d 2 x d d x M 2 =-f dt dx dt (3.7)
现代分离方法与技术-第3章:分离过程的动力学
如果定义: c Y =f d ext d + d x d x Y是外场和内部物理化学作用的总化学势产生的迁移
速度。将Y代入公式有: RT dc J =Yc (3.12) dx f 当外场和内部作用势能梯度为零(Y = 0)时,就只存在 扩散运动了,此时有: RT dc J = dx f 定义扩散系数D为: RT D= (3.13) f
现代分离方法与技术-第3章:分离过程的动力学
现代分离方法与技术-第3章:分离过程的动力学
如图3.1所示一种流体以非均匀线速度υ在x轴方向上流动。 当将流体分成很多层时,每一层的迁移速度是不同的。 对于相距dy的两个层而言,上层的移动要比下层快,由于 黏度的作用,上层流体的流速会因下层流体的流速慢而 趋于缓慢,同时下层流体会因上层流体的流速较快而趋 于加快流动。为了使这两层流体保持各自不变的流速,就 需要施加一个力F,单位面积上所感受到的这种力称为剪切 应力Fs,其大小与y轴方向上的流体流速梯度dυ/dy成正比。 F d 所以:Fs = = lim = y 0 y A dy (3.16)
现代分离方法与技术-第3章:分离过程的动力学
如上图所示,在流方向上取一小体积元,即厚度为dx 的截面。根据质量守恒定律,有: dm (J x - J x+dx )S= (3.31) dt 为简便起见,假设流方向上的截面积为一个单位,即 S = 1,并将式(3.31)右边分子分母同时乘dx后,得到: dm dx dx dc J x - J x+dx = =dc = dx (3.32) dx dt dt dt 将J x+dx在dx附近按Taylor级数展开,并略去高次项,得: J x+dx dJ J x + dx dx (3.33)
现代分离方法与技术-第3章:分离过程的动力学
宏观物体的机械运动规律可以用牛顿定律来描述。根据 牛顿运动规律,力F 与加速度的关系为: d2 x F = F1 + F2 = m 2 (3.3) dt 这个微分方程可以被积分并可表示物体在不同时间的瞬 d2 x 间位置。式中,m为物体的质量; 2 为加速度,F1为 dt 机械推动力,其大小由物体的位置决定,F2为摩擦力, dx 其大小与摩擦系数f 成比例,在运动速度较低时,与 dt 成正比,故以负号表示。
=A'T 1/2
等因素有关。
(3.27)
式中,A'为常数,与气体分子的质量和分子碰撞直径
现代分离方法与技术-第3章:分离过程的动力学
一种气体在另一种气体中的扩散取决于分子本身的迁移 性质,气体分子在浓度梯度作用下的迁移仍然遵守费克 第一扩散定律,只是不同的气体分子具有不同的扩散系 数而已。 低压下的混合气体可以近似看作理想气体,两种气体A和 B混合气体的扩散系数Dmix为: Dmix=xA DA +xB DB (3.28) 式中,x A和x B 分别为组分A和B的摩尔分数;D A和D B 分别为 A和B的扩散系数。对于刚性的球形分子而言,D A和D B为 0.599υλ,υ为分子运动的平均速度,λ为分子的麦克斯韦 (Maxwell)平均自由路径,式(3.28)可写成: Dmix=0.599(xAA A +xBBB ) (3.29)
现代分离方法与技术-第3章:分离过程的动力学
第3章 分离过程中的动力学
分子迁移-费克第一扩散定律 流体的迁移与扩散 带的迁移-费克第二扩散定律
有流存在下的溶质输运
现代分离方法与技术-第3章:分离过程的动力学
一个体系在达到平衡之前,体系内存在各种梯度,有外场
作用下的梯度,如压力梯度、浓度梯度、电位梯度和温度 梯度,也有体系内部的分子间相互作用引起的化学势梯度; 在分离过程中,溶质分子在外场或内部化学势作用下趋于 平衡的方向定向迁移,在空间上重新分配; 与此同时,溶质分子的随机运动又会使溶质分子从高浓度 区域向四周低浓度区域扩散,使分离开的溶质又趋向重新 混合; 定向迁移与非定向扩散,即分离与混合,是两种相伴而生 的趋势; 分离过程中动力学的研究内容就是物质在输运过程中运动 规律,即分离体系中组分迁移和扩散的基本性质和规律。现代分离方法与技术-第3Fra bibliotek:分离过程的动力学