(Ele 1999)advances in protein solubilisation for 2D electrophoresis
诺贝尔奖 突触囊泡.

2013诺贝尔奖】生理学奖深度解读:囊泡运输,细胞的“物流系统”Calo 2013-10-08 12:10一个细胞就好比一个人类社会。
人类社会有多复杂,细胞活动就有多精妙。
在日常生活中,我们需要进行有效率的生产生活,就必须有效率地调配生产资料与生活资源——因此,我们需要建立周密有效、安排得当的物流系统。
细胞也一样,基因的表达产物需要定位到不同的地点行使功能:膜蛋白需要奔向自己的靶位点、胰岛素需要分泌出细胞外、神经递质需要扩散到下一个神经细胞……要在正确的时间把正确的细胞货物运送到正确的目的地,细胞的物质转运机制之精妙,比无数物流师呕心沥血的杰作都更胜一筹。
而囊泡运输(vesicle trafficking),正是这一机制的重要组分。
2013年诺贝尔生理学或医学奖于10月7日颁布。
图片来源:昨天,2013年诺贝尔生理学或医学奖被授予发现囊泡转运机制的詹姆斯·罗斯曼、兰迪·谢克曼和托马斯·聚德霍夫3位科学家。
今天,让我们来看一下,大自然的物流师究竟是如何运筹于帷幄之中,决胜于细胞内外的呢?细胞中包括蛋白质在内的大多数分子都太大了,以致于不能直接穿过细胞中的膜结构。
于是,这些分子的运输需要依赖一种叫囊泡的细胞结构——这种有膜包被的小型泡状结构能够将待运输的分子包裹起来,送到目的地去释放掉。
可以想象,这种泡状的“集装箱”在运送细胞货物的过程中是极为重要的装备。
因此,在细胞中,尤其是在细胞质膜、内质网以及高尔基体中,囊泡的形成是持续不断的。
这些“集装箱”一旦被生产出来就马上投入使用,带着它们的货物奔向细胞内或细胞外的目的地。
囊泡之所以能够完成转运任务,是因为囊泡的膜与细胞质膜以及细胞内膜系统的组成成分是相似的,能够通过出芽的方式脱离转运起点、通过膜融合的方式归并到转运终点。
囊泡转运过程的第一步是膜通过出芽方式形成一个囊泡。
囊泡的外表面被蛋白包被。
通过改变膜结构的构象,这些蛋白将促使囊泡形成。
污水处理的英文文献翻译(1)

Nutrient removal in an A2O-MBR reactor with sludgereductionABSTRACTIn the present study, an advanced sewage treatment process has been developed by incorporating excess sludge reduction and phosphorous recovery in an A2O-MBR process. The A2O-MBR reactor was operated at a flux of 77 LMH over a period of 270 days. The designed flux was increased stepwise over a period of two weeks. The reactor was operated at two different MLSS range. Thermo chemical digestion of sludge was carried out at a fixed pH (11)and temperature (75℃) for 25% COD solubilisation. The released pbospborous was recovered by precipitation process and the organics was sent back to anoxic tank. The sludge digestion did not have any impact on COD and TP removal efficiency of the reactor. During the 270 days of reactor operation, the MBR maintained relatively constant transmembrane pressure. The results based on the study indicated that the proposed process configuration has potential to reduce the excess sludge production as well as it didn't detonated the treated water quality.Keywords: A2O reactor; MBR; Nutrient removal; TMP1. IntroductionExcess sludge reduction and nutrients removal are the two important problems associated with wastewater treatment plant. MBR process has been known as a process with relatively high decay rate and less sludge production due to much longer sludge age in the reactor (Wenet al., 2004). Sludge production in MBR is reduced by 28-68%, depending on the sludge age used (Xia et al.,2008). However, minimizing the sludge production by increasing sludge age is limited due to the potential adverse effect of high MLSS concentrations on membrane (Yoon et al., 2004). This problem can be solved by introducing sludge disintegration technique in MBR (Young et al., 2007). Sludge disintegration techniques have been reported to enhance the biodegradability of excess sludge (Vlyssides and Karlis, 2004). In overall, the basis for sludge reduction processes is effective combination of the methods for sludge disintegration and biodegradation of treated sludge. Advances in sludge disintegration techniques offer a few promising options including ultrasound (Guo et al., 2008), pulse power (Choi et al.,2006), ozone (Weemaes et al., 2000), thermal (Kim et al., 2003), alkaline (Li et al., 2008) acid (Kim et al., 2003) and thermo chemical(Vlyssides and Karlis, 2004). Among the various disintegration techniques, thermo chemical was reported to be simple and cost effective (Weemaes and Verstraete, 1998). In thermal-chemical hydrolysis, alkali sodium hydroxide was found to be the most effective agent in inducing cell lysis (Rocker et al., 1999). Conventionally, the nutrient removal was carried out in an A2O process. It has advantage of achieving, nutrient removal along with organic compound oxidation in a single sludge configuration using linked reactors in series (Tchobanoglous et al., 2003). The phosphoroes removal happens by subjecting phosphorous accumulating organisms (PAO) bacteria under aerobic and anaerobic conditions (Akin and Ugurlu, 2004). These operating procedures enhance predominance PAO, which are able to uptake phosphorous in excess. During the sludge pretreatment processes the bound phosphorous was solubilised and it increases the phosphorousconcentration in the effluent stream (Nishimura, 2001).So, it is necessary to remove the solubilised phosphorus before it enters into main stream. Besides, there is a growing demand for the sustainable phosphorous resources in the industrialized world. In many developed countries, researches are currently underway to recover the phosphoroes bound in the sludge's of enhanced biological phosphorus removal system (EBPR). The released phosphorous can be recovered in usable products using calcium salts precipitation method. Keeping this fact in mind, in the present study, a new advanced wastewater treatment process is developed by integrating three processes, which are: (a) thermo chemical pretreatment in MBR for excess sludge reduction (b) A2O process for biological nutrient removal (c) P recovery through calcium salt precipitation. The experimental data obtained were then used to evaluate the performance of this integrated system.2. Methods2.1. WastewaterThe synthetic domestic wastewater was used as the experimental influent. It was basically composed of a mixed carbon source, macro nutrients (N and P), an alkalinity control (NaHCO3) and a microelement solution. The composition contained (/L) 210 mg glucose, 200 mg NH4C1, 220 mg NaHCO3, 22一34 mg KH2PO4, microelement solution (0.19 mg MnCl2 4H20, 0.0018 mg ZnCl22H2O,0.022 mg CuCl22H2O, 5.6 mg MgSO47H2O, 0.88 mg FeCl36H2O,1.3 mg CaCl2·2H2O). The synthetic wastewater was prepared three times a week with concentrations of 210±1.5 mg/L chemical oxygen demand (COD), 40±1 mg/L total nitrogen (TN) and 5.5 mg/L total phosphorus (TP).2.2. A2O-MBRThe working volume of the A2O-MBR was 83.4 L. A baffle was placed inside the reactor to divide it into anaerobic (8.4 L) anoxic (25 L) and aerobic basin (50 L). The synthetic wastewater was feed into the reactor at a flow rate of 8.4 L/h (Q) using a feed pump. A liquid level sensor, planted in aerobic basin of A2O-MBR controlled the flow of influent. The HRT of anaerobic, anoxic and aerobic basins were 1, 3 and 6 h, respectively. In order to facilitate nutrient removal, the reactor was provided with two internal recycle (1R). IRl (Q= 1)connects anoxic and anaerobic and IR 2 (Q=3) was between aerobic and anoxic. Anaerobic and anoxic basins were provided with low speed mixer to keep the mixed liquid suspended solids (MLSS) in suspension. In the aerobic zone, diffusers were used to generate air bubbles for oxidation of organics and ammonia. Dissolved oxygen (DO) concentration in the aerobic basin was maintained at 3.5 mg/1 and was monitored continuously through online DO meter. The solid liquid separation happens inaerobic basin with the help of five flat sheet membranes having a pore size of 0.23 pm. The area of each membrane was 0.1 m2. They were connected together by a common tube. A peristaltic pumpwas connected in the common tube to generate suction pressure. In the common tube provision was made to accommodate pressure gauge to measure transmembrane pressure (TMP) during suction. The suction pump was operated in sequence of timing, which consists of 10 min switch on, and 2 min switch off.2.3. Thermo chemical digestion of sludgeMixed liquor from aerobic basin of MBR was withdrawn at the ratio of 1.5% of Q/day and subjected to thermo chemical digestion. Thermo chemical digestion was carried out at a fixed pH of 11(NaOH) and temperature of 75℃for 3 h. After thermo chemical digestion the supernatant and sludge were separated. The thermo-chemicallydigested sludge was amenable to further anaerobic bio-degradation (Vlyssides and Karlis, 2004), so it was sent to theanaerobic basin of the MBR2.4. Phosphorus recoveryLime was used as a precipitant to recover the phosphorous in the supernatant. After the recovery of precipitant the content was sent back to anoxic tank as a carbon source and alkalinity supelement for denitrification.2.5. Chemical analysisCOD, MLSS, TP, TN of the raw and treated wastewater were analyzed following methods detailed in (APHA, 2003). The influent and effluent ammonia concentration was measured using an ion-selective electrode (Thereto Orion, Model: 95一12). Nitrate in the sample was analyzed using cadmium reduction method (APHA, 2003).3. Results and discussionFig. 1 presents data of MLSS and yield observed during the operational period of the reactor. One of the advantages of MBR reactor was it can be operated in high MLSS concentration. The reactor was seeded with EBPR sludge from the Kiheung, sewage treatment plant, Korea. The reactor was startup with the MLSS concentration of 5700 mg/L. It starts to increase steadily with increase in period of reactor operation and reached a value of 8100 mg/L on day 38. From then onwards, MLSS concentration was maintained in the range of 7500 mg/L by withdrawing excess sludge produced and called run I. The observed yields (Yobs) for experiments without sludge digestion (run I) and with sludge digestion were calculated and given in Fig. 1. The Yobs for run I was found to be 0.12 gMLSS/g COD. It was comparatively lower than a value of 0.4 gMLSS/g CODreported for the conventional activated sludge processes (Tchoba-noglous et al., 2003). The difference in observed yield of these two systems is attributed to their working MLSS concentration. At high MLSS concentration the yield observed was found to be low (Visva-nathan et al., 2000). As a result of that MBR generated less sludge.The presently used MLSS ranges (7.5一10.5 g/L) are selected on the basis of the recommendation by Rosenberger et al. (2002). In their study, they reported that the general trend of MLSS increase on fouling in municipal applications seems to result in no impact at medium MLSS concentrations (7一12 g/L).It is evident from the data that the COD removal efficiency of A2O system remains unaffected before and after the introduction of sludge digestion practices. A test analysis showed that the differences between the period without sludge digestion (run I) and with sludge digestion (run II and III) are not statistically significant.However, it has been reported that, in wastewater treatment processes including disintegration-induced sludge degradation, the effluent water quality is slightly detonated due to the release of nondegradable substances such as soluble microbial products (Ya-sui and Shibata, 1994; Salcai et al., 1997; Yoon et al., 2004). During the study period, COD concentration in the aerobic basin of MBR was in the range of 18-38 mg/L and corresponding organic concentration in the effluent was varied from 4 to 12 mg/L. From this data it can be concluded that the membrane separation played an important role in providing the excellent and stable effluent quality.Phosphorus is the primary nutrient responsible for algal bloom and it is necessary to reduce the concentration of phosphorus in treated wastewater to prevent the algal bloom. Fortunately its growth can be inhibited at the levels of TP well below 1 mg/L (Mer-vat and Logan, 1996).Fig. 2 depicts TP removal efficiency of the A2O-MBR system during the period of study. It is clearly evident from the figure that the TP removal efficiency of A/O system was remains unaffected after the introduction of sludge reduction. In the present study, the solubilised phosphorous was recovered in the form of calcium phosphate before it enters into main stream. So, the possibility of phosphorus increase in the effluent due to sludge reduction practices has been eliminated. The influent TP concentration was in the range of 5.5 mg/L. During thefirst four weeks of operation the TP removal efficiency of the system was not efficient as the TP concentration in the effluent exceeds over 2.5 mg/L. The lower TP removal efficiency during the initial period was due to the slow growing nature of PAO organisms and other operational factors such as anaerobic condition and internal recycling. After the initial period, the TP removal efficiency in the effluent starts to increase with increase in period of operation. TP removal in A2O process is mainly through PAO organisms. These organisms are slow growing in nature and susceptible to various physicochemical factors (Carlos et al., 2008). During the study period TP removal efficiency of the system remains unaffected and was in the range of 74-82%.。
年终盘点:2016年Nature杂志重磅级突破性研究成果

年终盘点:2016年Nature杂志重磅级突破性研究成果小福利:点击上方图片下载生物谷APP轻松获得本文中所有文献原文!年终专题系列——Nature杂志重磅研究时间过得总是很快,2016年已经步入尾声,迎接我们的将是崭新的2017年!在2016年里三大国际著名杂志Cell、Nature和Science(CNS)依然刊登了很多非常耐人寻味的研究成果,本文中谷君就盘点了2016年Nature杂志发表的一些突破性的重磅级研究,分享给大家!【1】Nature:中国首次利用CRISPR–Cas9编辑过的细胞开展人体临床试验doi:10.1038/nature.2016.20988来自中国成都市四川大学华西医院的一个研究人员团队首次将利用CRISPR–Cas9进行过基因编辑的细胞注射到一名病人体内。
《自然》期刊报道这一注射过程是在2016年10月28日发生的,而且迄今为止,这名病人表现得“还不错”。
经过基因修饰的细胞之前已被注射到人体内,但是是利用不同的技术实现的。
CRISPR-Cas9被认为是一种更加高效的方法。
在这项新的努力中,该团队从血液样品中分离出免疫细胞,然后利用CRISPR-Cas9寻找它们中的PD-1蛋白,并且让该蛋白不能发挥功能,而之前的研究已证实这会延缓免疫细胞作出的免疫反应。
人们的看法是让这种蛋白失去功能将允许免疫系统更强地抵抗肿瘤生长。
这些利用CRISPR-Cas9进行过基因编辑的细胞被放置在一个容器中,在那里,它们在体外培养后能够发生增殖---它们随后经收集后被注射到一名肺癌病人体内,其中这名病人已不能够对任何其他的疗法作出反应。
这种CRISPR-Cas9技术涉及利用一种结合特定DNA序列的向导RNA和一种能够在事先选择的位点上切割DNA链的Cas9酶,从而允许移除DNA链,或者加入新的DNA片段。
【2】Nature:实验性疫苗与免疫刺激剂组合使用有望治疗HIV doi:10.1038/nature20583在一项新的研究中,来自美国贝斯以色列女执事医疗中心、沃尔特里德陆军研究院、Janssen疫苗与预防公司(Janssen Vaccines & Prevention B.V.)和吉利德科学公司的研究人员证实将一种实验性疫苗与一种先天性免疫刺激剂结合在一起可能有助导致HIV感染者体内的病毒缓解。
利用BiFC技术研究柔嫩艾美耳球虫顶膜抗原1结构域Ⅰ与棒状体颈部蛋

利用BiFC 技术研究柔嫩艾美耳球虫顶膜抗原1结构域互作关系严 茗1,2,黄 兵22,吕 凌1,2,董 辉2(1.上海师范大学生命与环境科学学院,上海 200234;2.中国农业科学院上海兽医研究所 农业部动物寄生虫学重摘 要:为了验证柔嫩艾美耳球虫(Eimeria tenella )顶膜抗原1结构域Ⅰ(apical membrance antigen 1 domain I ,Et AMA1-D Ⅰ)与棒状体颈部蛋白2(rhoptry neck protein 2,Et RON2)的互作关系,以柔嫩艾美耳球虫子孢子cDNA 为模板,PCR 扩增出 492 bp 的Et AMA1-D Ⅰ片段和1395 bp 的Et RON2片段,并与pGEM-T-easy 载体连接构建相应重组质粒。
获得的阳性重组质粒及双分子荧光互补技术的真核表达载体pBiFC-VN155和pBiFC-VC155用Eco R Ⅰ和BgI II 进行双酶切,将Et AMA1-D Ⅰ、Et RON2分别与pBiFC-VN155、pBiFC-VC155连接,构建真核重组质粒pBiFC-VN155-Et AMA1-D Ⅰ和pBiFC-VC155-Et RON2。
将2个真核重组质粒分别转染BHK 细胞进行表达,经间接免疫荧光鉴定,可在BHK 细胞中成功表达。
将2个真核重组质粒共转染至BHK 细胞中,同时将pBiFC-bJunVN155和pBiFC-bFos(deltaZIP)VC155、pBiFC-bJunVN155和pBiFC-bFos(delta)VC155共转染至细胞中分别作为阳性和阴性对照组。
BiFC 结果发现真核重组质粒共转染组和阳性对照组的BHK 细胞均产生绿色荧光,而阴性对照组无荧光,表明Et AMA1-D Ⅰ与Et RON2蛋白之间存在互作关系。
本研究结果为深入研究Et AMA1及Et RON2在球虫入侵过程中的功能与作用机制奠定基础。
1GGH安装作业指导书005 (613)
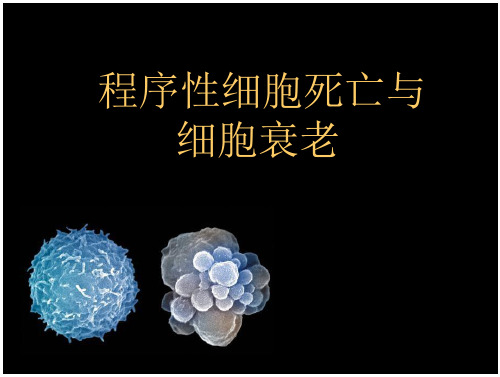
细细胞胞凋亡凋亡和细胞坏死细的胞坏区死
生理性、温和刺激或药物 非同步性、散在、单个细胞
别 病理性、剧烈刺激、损伤
大块组织同时发生
细胞体积缩小
细胞膨胀(内质网、线粒体等)
执行caspase 死亡受体途径的起始caspase 起始caspase
Caspase-12
死亡受体途径的起始caspase
IL-1前体的切割死亡受体途径的起始caspase
Caspase有两类:一类是凋亡起始者,包括 caspase2、caspase8、caspase9;一类是凋亡 执行者,包括caspase3、caspase6.caspase7 起始者负责对执行者的前体进行切割,从而产 生有活性的执行者;执行者负责切割细胞核内 、细胞质中的结构蛋白和调节蛋白。。
Robert Horvitz(罗伯特• 霍维茨)发现了C. elegan中控 制细胞死亡的关键基因(功能性ced-3 和ced-4基因是细 胞死亡执行的一种先决条件, ced-9通过与ced- 3 和ced-4 相互作用阻止细胞死亡), 并描述了其特征他显示了这些 基因在细胞死亡过程中如何互相作用, 并揭示相应的基因 也存在于人体之中。。
Caspase活化
细胞凋亡过程可分为激活期和执行期两个阶段。 前期细胞应答死亡信号, 起始caspase活化;后期 执行caspase活化, 执行细胞死亡程序。 Caspase自身以非活化的Procaspase存在于细胞 质中, 其激活依赖于其他的Caspase在它的天冬氨 酸位点裂解活化或自身活化。 起始Caspase的活化属于同性活化, 执行Caspase 的活化属于异性活化。。
原始纯化蛋白质文献

RESEARCHA number of systems have been developed to simplify protein purifica-tion through the expression of recombinant proteins as fusions to car-rier proteins or peptides 1,2. Examples of commercially available systems include maltose binding protein 3, glutathione S -transferase 4, biotin carboxyl carrier protein 5,6, thioredoxin 6,7, and cellulose binding domain 8. Similarly, vectors for fusion to short peptide tags such as oligohistidine 9, S-peptide 10, and the FLAG tm peptide 11are also available.These systems typically allow one-step purification of a recombinant protein from cell extract by affinity chromatography, using an immobi-lized moderate-affinity ligand specific to the carrier protein 12.Although it is useful for laboratory-scale purification, affinity chro-matography on scale-up can represent a major cost of the final protein product at the preparative scale. More economical and technically sim-ple methods for purification of soluble proteins are therefore desirable.Elastin-like polypeptides (ELPs) are oligomeric repeats of the pentapeptide Val-Pro-Gly-Xaa-Gly (where the “guest residue” Xaa is any amino acid with the exception of proline) that undergo a reversible inverse temperature transition. They are highly soluble in water below the inverse transition temperature (T t ), but undergo a sharp (2–3°C range) phase transition when the temperature is raised above T t , leading to desolvation and aggregation of the polypep-tide 13–15. In previous work, McPherson et al. exploited the inverse transition to purify recombinant poly(Gly-Val-Gly-Val-Pro)polypeptides 16.Previous studies have also shown that protein conju-gates of poly(N -isopropylacrylamide), a synthetic polymer that undergoes a similar thermally reversible phase transition, also retain the transition behavior of the free polymer 17–19. Together, these observations suggested that the thermally driven inverse transition of an ELP is likely to be maintained upon incorporation in a fusion protein.We hypothesized that, upon incorporation in fusion pro-teins, ELPs would allow nonchromatographic, thermally stimulated phase separation of recombinant proteins.ResultsWe sought to design an ELP sequence with a predicted T t >37°C, so that the ELP fusion protein would remain soluble during culture in Escherichia coli,but aggregate in response to a small increase in tem-perature. The composition and chain length of guest residue(s) have been shown to affect the T t strongly 20,21. We synthesized a gene encod-ing an ELP with guest residues Val, Ala, and Gly in the ratio 5:2:3 that had a predicted T t of approximately 40°C in water for a molecular mass (M r ) greater than 50 kDa. The synthetic gene encoded 10 Val-Pro-Gly-Xaa-Gly repeats (the 10-mer or ELP10) and was oligomer-ized up to 18 times to create a library of genes encoding ELPs with pre-cisely specified molecular masses ranging from 3.9 to 70.5 kDa. T o our knowledge, these are the first examples of genetically engineered ELPs of precisely defined chain length and amino acid sequence that are designed to exhibit an inverse transition at a specified temperature.Genes for two other proteins were fused to the ELP genes. First,each ELP sequence (10-, 20-, 30-, 60-, 90-, 120-, 150-, and 180-mers) was fused to the C terminus of E. coli thioredoxin, a 109-residue protein that is commonly used as a carrier to increase the solubility of recombinant proteins 7. Second, tendamistat, a 77-residue protein inhibitor of α-amylase 22, was fused to the C terminus of a thioredoxin–ELP90 fusion, forming a ternary fusion. The ELP fusion proteins were expressed in E. coli and purified from cell lysate by either immobilized metal affinity chromatography (IMAC)23or inverse transition cycling (described below). The purified ELP fusion protein was cleaved with thrombin to release the target pro-tein from the ELP , which was separated from the target protein by an additional round of inverse transition cycling. For each construct,the purified ELP fusion protein, target protein, and ELP were char-acterized for size and purity by SDS–PAGE (results not shown).The inverse transition can be monitored by assaying solution turbidity spectrophotometrically as a function of temperature.Increase in temperature beyond a critical point results in a sharp increase in turbidity over an approximately 2°C range to a maxi-mum value (OD 350approximately 2.0), because of aggregation of the ELP . The T t , defined as the temperature at 50% maximal turbidity, is a convenient parameter to describe this process. The inverse transi-tion profiles of free ELP , thioredoxin–ELP fusion, ELP–tendamistat fusion, and ternary thioredoxin–ELP–tendamistat fusion in phos-phate-buffered saline (PBS) are shown in Figure 1A. The T t was 51°C for free ELP and 54°C for the thioredoxin fusion, showing that the T tPurification of recombinant proteins by fusion with thermally-responsivepolypeptidesDan E. Meyer and Ashutosh Chilkoti*Department of Biomedical Engineering, Box 90281, Duke University, Durham, NC 27708-0281. *Corresponding author (chilkoti@).Received 2 June 1999; accepted 30 July 1999Elastin-like polypeptides (ELPs) undergo a reversible, inverse phase transition. Below their transition temperature (T t ), ELPs are soluble in water, but when the temperature is raised above T t , phase transition occurs, leading to aggregation of the polypeptide. We demonstrate a method for purification of soluble fusion proteins incorporating an ELP tag. Advantages of this method, termed “inverse transition cycling,”include technical simplicity, low cost, ease of scale-up, and capacity for multiplexing. More broadly, the ability to environmentally modulate the physicochemical properties of recombinant proteins by fusion with ELPs will allow diverse applications in bioseparation, immunoassays, biocatalysis, and drug delivery.Keywords:elastin-like polypeptide, inverse phase transition, environmentally responsive, fusion protein, protein purification© 1999 N a t u r e A m e r i c a I n c . • h t t p ://b i o t e c h .n a t u r e .c o mRESEARCHis only slightly affected by fusion to thioredoxin. Thioredoxin–ELP produced by cleavage from the ternary tendamistat fusion had a higher T t compared with thioredoxin–ELP produced directly (60versus 54°C), presumably because of differences in the leading and trailing amino acid sequences immediately adjacent to the ELP sequence (see Fig. 3). Transition profiles of ELP–tendamistat and thioredoxin–ELP–tendamistat fusion proteins were nearly identical,with a T t of 34°C. Aggregation of the ELP fusion proteins was reversible, and the aggregates were resolubilized completely when the temperature was lowered below the T t . Thioredoxin and ten-damistat controls exhibited no change in absorbance with increasing temperature, indicating that the induced aggregation observed for the fusion proteins was due to the inverse transition of the ELP carri-er. Typically, the inverse transition of the fusion proteins was also slightly broader than that of free ELP , with small upper and lower shoulders observed in their turbidity profiles.Motivated by the studies of Urry et al.21, who observed a decrease in T t with increasing chain length, we also investigated the effect of ELP molecular mass on the inverse transition of ELP fusion proteins.The T t of a set of thioredoxin–ELP fusion proteins was determined as a function of the molecular mass of the ELP carrier, which ranged from 11.8 to 70.5 kDa (Fig. 1B). The T t values for the higher molecu-lar mass fusion proteins approached the design target temperature of 40°C, whereas the T t values for the lower molecular mass fusions were significantly greater.In addition to ELP-specific variables that affect the T t (i.e., guest residue composition and M r ), several extrinsic factors can further modulate the T t for a given ELP . Such factors include the choice of sol-vent, ELP concentration, and ionic strength 15. Controlling the ionic strength, in particular, allows the T t to be tuned over a 50°C range (Fig.1C), and thereby provides a means for isothermal triggering of the transition. The T t increases with decreasing ELP concentration, and for concentrations less than about 25 µM this effect becomes significant 22.However, this effect can be mitigated by adding salt to depress the T t ,thereby allowing recovery of ELP fusion proteins from dilute solutions.The specific activity of the thioredoxin–ELP60 fusion protein,determined by an insulin reduction assay, was identical to that of com-mercial E. coli thioredoxin (results not shown), indicating that below the T t , the ELP tag had no effect on thioredoxin activity. We also mon-itored thioredoxin activity during thermal transition cycling, in which a sample of thioredoxin–ELP60 was cycled between temperatures above and below the T t (results not shown). When resolubilized below the T t , no change in activity was observed, even after four such cycles,which confirmed that the temperature changes and resulting aggrega-tion/resolubilization had no effect on stability and function of the fused target protein. For the ternary thioredoxin–ELP–tendamistat fusion, an α-amylase inhibition assay showed that the thioredoxin–ELP90 carrier reduced the inhibitory activity of tendami-stat by twofold (results not shown). However, after thrombin cleavage and purification of tendamistat from the thioredoxin-ELP carrier, the activity of purified tendamistat was indistinguishable from recombi-nant tendamistat that was independently purified by IMAC.Six thioredoxin–ELP fusion proteins containing a C-terminal 30,60-, 90-, 120-, 150-, or 180-mer ELP tag, and the thioredoxin-ELP90–tendamistat fusion protein were purified from cell lysate by inverse transition cycling, achieved by repeated centrifugation at conditions (i.e., NaCl concentration and temperature) alternating above and below T t . Typical results are shown in Figure 2A. Figure 2B shows the corresponding thioredoxin activity and total protein at each stage of purification of the thioredoxin–ELP fusion. Before purification, the induced E. coli were harvested from culture media by centrifugation, resolubilized in a low-salt buffer, and lysed by ultrasonic disruption. After high-speed centrifugation to remove insoluble matter, polyethylenimine was added to the lysate to precip-Figure 1. Characterization of the inverse transition of ELP fusion proteins. (A) Turbidity profiles for free ELP (thrombin-cleaved and purified from thioredoxin–ELP) (v ); thioredoxin–ELP (̆); thioredoxin–ELP–tendamistat (ć); ELP–tendamistat (cleaved and purified from thioredoxin-ELP-tendamistat) (ȣ); and thioredoxin–ELP (cleaved and purified from thioredoxin–ELP–tendamistat) ( ). All fusion proteins contained the same 90-pentapeptide ELP , with 25 µM protein concentration in PBS using a 1.5°C min -1heating rate. (B) T t as a function of ELP M r for the thioredoxin–ELP fusion proteins. (C) T t as a function of NaCl concentration for the thioredoxin/60-mer ELP fusion protein (25 µM) in 50 mM phosphate buffer, pH 8.0.Figure 2. Purification by inverse transition cycling. (A)SDS–PAGE of each stage of purification for the thioredoxin–ELP90fusion (49.9 kDa, lanes 1–5) and the thioredoxin–ELP90–tendamistat (57.4 kDa,lanes 7–9). Lanes 1 and 7, soluble lysate;lanes 2 and 8, supernatant containing contaminating E. coli proteins; lanes 3 and 9, resolubilized pellet containing purified fusion protein; lane 4, second-round supernatant;lane 5, second-round pellet;lanes 6 and 10: Molecular mass markers (kDa). (B) Total protein (by bicinchonic acid assay) and thioredoxin activity (by insulin reduction assay) for each stage of purification (lanes 1–5 as in part A) of the thioredoxin–ELP90. Values were normalizedto the soluble lysate.A BA B C© 1999 N a t u r e A m e r i c a I n c . • h t t p ://b i o t e c h .n a t u r e .c o mRESEARCHitate nucleic acids, yielding soluble lysate (lanes 1 and 7). Inverse transition cycling was initiated by adding NaCl and/or increasing the solution temperature to induce the ELP inverse transition, causing the solution to become turbid as a result of aggregation of the ELP fusion protein. The aggregated fusion protein was separated from solution by centrifugation at a temperature greater than the T t , and a translucent pellet formed at the bottom of the centrifuge tube. The supernatant, containing contaminating E. coli proteins comprising approximately 80% of the total protein, was decanted and discarded(lanes 2 and 8). The low thioredoxin activity measured in the super-natant, a portion of which is contributed by native E. coli thioredox-in, confirmed that this fraction primarily contained contaminatinghost proteins and not the overexpressed thioredoxin–ELP fusionprotein. The pellet was redissolved in a buffer of low ionic strength ata temperature below T t and centrifuged at low temperature to remove any remaining insoluble matter (lanes 3 and 9). The thiore-doxin-specific activity of the resolubilized protein approached thatof commercially available thioredoxin (data not shown). Althoughadditional rounds of inverse transition cycling were undertaken(lanes 4 and 5), the level of contaminating proteins was below detec-tion after a single round of inverse transition cycling, and nodetectable increase in thioredoxin-specific activity was obtained.Protein yields for the thioredoxin fusion constructs were typical-ly >40 mg of purified fusion protein per liter of culture. We foundthat the total gravimetric yield of fusion protein decreased withincreasing ELP length, with the 30-mer averaging about 70 mg/L andthe 180-mer averaging about 40 mg/L. However, ELP length appearsto have no effect on purification efficiency, and recovery from celllysate was complete for all fusion constructs. Expression levels of sol-uble tendamistat were slightly larger for the ternarythioredoxin–ELP–tendamistat fusion (40 mg/L ternary fusion, or 6mg/L tendamistat) compared with fusion with thioredoxin only (10mg/L thioredoxin–tendamistat fusion, 4 mg/L tendamistat).DiscussionWe chose thioredoxin and tendamistat as target proteins because theyexemplify two extremes of soluble protein expression. Thioredoxin isoverexpressed at high levels and is highly soluble, and tendamistat isexpressed largely as insoluble inclusion bodies. Proteins representa-tive of the latter class, however, must exhibit some level of solubleexpression to be purified by inverse transition cycling. Althoughfusion with thioredoxin is known to promote the soluble expressionof target proteins 7, we have observed that only 5–10% of overex-pressed thioredoxin–tendamistat fusion protein is recovered as solu-ble and functionally active protein (A.C., unpublished data).The thioredoxin–ELP fusion exhibits only a small increase in T t (1–2°C) com-pared with free ELP , whereas the tendamis-tat fusion displayed a dramatic 25°C reduction in T t . This shift is identical for both the ternary (thioredoxin–ELP–ten-damistat) and binary (ELP–tendamistat)constructs, indicating that the shift in T t is associated specifically with tendamistat.We hypothesize that the observed decrease in T t is due to interactions between the ELPchain and solvent-exposed hydrophobicregions in tendamistat, whereas for the highly soluble thioredoxin, these hydrophobic interactions are negligible.Although this T t shift adds complexity to the design of ELP carriers for inverse tran-sition purification of proteins containing a large exposed hydrophobic area, for highly soluble proteins only a small perturbationof T t relative to the free ELP is likely to be introduced upon fusion with an ELP tag. In addition to surface hydrophobicity, variability in target protein size is another factor that might be expected to affect the effi-ciency of purification by inverse transition cycling. Although the results reported here are for small target proteins (<20 kDa), we have also purified immune complexes with a total molecular mass greater than 230 kDa (D.E.M. and A.C., unpublished data).The ELP M r has two important ramifications for fusion proteinpurification. In buffers of low ionic strength, the T t values of the lower M r ELPs are too high for purification and would require a high concentration of NaCl to decrease to a useful temperature.The ELP chain length is also important with respect to protein yields. In addition to the decreased total yield of fusion protein observed with increasing ELP M r , the mass percentage of target protein is further decreased. Because we have observed that purifi-cation efficiency does not appear to be reduced for lower M r ELPs,the design of our next generation ELP carriers will maximize target protein expression by minimizing the ELP M r , while retaining a target T t of about 40°C through the incorporation of a larger frac-tion of hydrophobic guest residues in the ELP sequence.Inverse transition cycling offers several advantages for purifica-tion of recombinant fusion proteins. First, the expense associated with chromatographic resins and equipment is eliminated. The potentially lower cost and ease of scale-up of this method are likely to prove attractive for large-scale (in the grams to kilograms range)purification of proteins. Second, the separation and recovery condi-tions are mild, requiring only a modest change in temperature or ionic strength. Third, the method is fast and straightforward, with only a few short centrifugation or filtration steps followed by resolu-bilization of the purified protein in a buffer of low ionic strength.Fourth, inverse transition cycling can be easily multiplexed to purify proteins simultaneously from different cell cultures. Because protein purification is frequently the limiting step in structure/function studies and in screening of proteins in pharmaceutical development,we believe that the ability to purify simultaneously a large number of proteins by inverse transition purification is likely to be extremely useful for laboratory-scale purification (in the microgram to mil-ligram range) of proteins. Finally, inverse transition purification is independent of a specific expression vector or host and is likely to be extremely useful for eukaryotic expression systems because of their ability to overexpress heterologous proteins in a soluble state 24.In addition to protein purification, a number of different appli-cations for protein-ELP conjugates can be envisioned. Enzyme–ELP fusions could be useful substitutes for immobilized enzymes inindustrial biocatalysis, by allowing easy separation of the enzymeFigure 3. Gene sequences. (A) The 10-pentapeptide ELP gene. (B) The modified pET -32b vector forproduction of thioredoxin–ELP fusions. (C) The modified pET -32a vectors for the production of thethioredoxin–ELP–tendamistat fusions with alternate thrombin cleavage sites.ABC © 1999 N a t u r e A m e r i c a I n c . • h t t p ://b i o t e c h .n a t u r e .c o mRESEARCHfrom product and recycling of the enzyme for subsequent rounds of biocatalysis 25. Similarly, fusion of ELPs with engineered antibodies could allow the capture of an analyte from biological fluids for immunoassays 26. The use of ELPs conjugated to radionuclides or protein therapeutics for precise targeting for imaging and therapy by targeted hyperthermia 27–29is also currently under investigation.Experimental protocolGene synthesis.Standard molecular biology protocols were used for gene synthesis and oligomerization 30. All clones were maintained in the XL1-Blue strain of E. coli . Colonies were screened by PCR, and the DNA sequence of all putative inserts was verified by automated fluorescent dye terminator DNA sequencing.A synthetic gene for a 10-polypentapeptide ELP was constructed from four 5′-phosphorylated, PAGE-purified synthetic oligonucleotides (Integrated DNA Technologies , Coralville, IA). The oligonucleotides were annealed to form double-stranded DNA with Eco RI- and Hin dIII-compati-ble ends (Fig. 3A), and ligated into Eco RI/Hin dIII linearized and dephospho-rylated pUC-19 (N. Engl. Biolabs, Beverly, MA). The gene sequence was selected to emphasize E. coli preferred codons while also minimizing sequence repetition. For a typical oligomerization, the vector was linearized with Pfl MI and enzymatically dephosphorylated. The insert was doubly digested with Pfl MI and Bgl I, purified by agarose gel electrophoresis (QIAquick Gel Extraction Kit, Qiagen, Valencia, CA), and ligated into the lin-earized vector.Fusion protein construction and expression . For the thioredoxin fusions,pET-32b (Novagen, Madison, WI)was modified to contain an Sfi I restric-tion site downstream of the thioredoxin gene (Fig. 3B). For the ternary ten-damistat fusion, a pET-32a-based plasmid containing a gene for a thiore-doxin–tendamistat fusion was modified to contain an Sfi I restriction site in two alternate locations, upstream or downstream of the thrombin recogni-tion site (Fig. 3C). The ELP gene segments, produced by digestion with Pfl MI and Bgl I, were then ligated into the Sfi I site of each modified expres-sion vector.Expression vectors were transformed into the expression strains BLR(DE3) (thioredoxin) or BL21-trxB(DE3) (tendamistat) (Novagen).Shaker flasks with CircleGrow media (Bio 101,La Jolla, CA), supplemented with 100 µg/ml ampicillin, were inoculated, incubated at 37°C with shaking (250 r.p.m.), and induced at an OD 600of 0.8 by the addition of isopropyl α-thiogalactopyranoside (1 mM final concentration). The cultures were incu-bated an additional 3 h, harvested by centrifugation at 4°C, resolubilized in low-ionic-strength buffer (∼1/30 culture volume), and lysed by ultrasonic disruption at 4°C. The lysate was centrifuged at ∼20,000 g at 4°C for 15 min to remove insoluble matter. Nucleic acids were precipitated by the addition of polyethylenimine (0.5% final concentration), followed by centrifugation at ∼20,000 g at 4°C for 15 min.Fusion protein purification. The thioredoxin fusions, which contained a (His)6tag, were purified by IMAC using a nickel-chelating nitrilotriacetic-derivatized resin (Novagen)23or, alternatively, by inverse transition cycling.The tendamistat fusion was purified exclusively by inverse transition cycling.For purification by inverse transition cycling, ELP fusion proteins were aggregated by increasing the temperature of the cell lysate to ≤45°C and/or by adding NaCl to a concentration ≤2 M. The aggregated fusion protein was sep-arated from solution by centrifugation at 35–45°C at 10,000–15,000 g for 15min. The supernatant was decanted and discarded, and the pellet containing the fusion protein was resolubilized by agitation in cold, low-ionic-strength buffer. The resolubilized pellet was then centrifuged at 4°C to remove any remaining insoluble matter.Characterization of ELP fusion proteins. The optical absorbance at 350nm of ELP fusion solutions were monitored in the 4–80°C range on a Cary 300 ultraviolet-visible spectrophotometer equipped with a multicell thermo-electric temperature controller (Varian Instruments, Walnut Creek, CA). The T t was determined from the midpoint of the transition-induced change at a heating or cooling rate of 1.5°C min -1. The SDS–PAGE analysis used precast Mini-PROTEAN 10–20% gradient gels (Bio-Rad, Hercules, CA) with a dis-continuous buffer system 24, stained with Coomassie brilliant blue. The con-centration of the fusion proteins was determined spectrophotometrically using calculated extinction coefficients. Total protein concentrations were determined by bicinchonic acid assay (Pierce Chemical Co., Rockford, IL).Thioredoxin activity was determined using an insulin reduction colorimet-ric assay 31. Tendamistat activity was determined by a colorimetric α-amy-lase inhibition assay (Sigma, St. Louis, MO).AcknowledgmentsWe thank R.T. Piervincenzi for the thioredoxin–tendamistat gene. This work was supported by Duke University through its provision of start-up funds to A.C., by the Whitaker Foundation, the North Carolina Biotechnology Center (ARIG no. 9605-ARG-0050), and the National Institutes of Health (1R21-GM-057373-01). We also thank the Whitaker Foundation for support of D.E.M. as a graduate fellow.1. Nilsson, B., Forsberg, G., Moks, T., Hartmanis, M. & Uhlén, M. Fusion proteins in biotechnology and structural biology. Curr. Opin. Struct. Biol.2, 569–575 (1992).2. Uhlén, M. & Moks, T. Gene fusions for purpose of expression: an introduction.Methods Enzymol.195, 129–143 (1990).3. Maina, C.V . et al. An Escherichia coli vector to express and purify foreign proteins by fusion to and separation from maltose binding protein. Gen e 74, 365–373(1988).4. Smith, D.B. & Johnson, K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase.Gen e 67, 31–40(1988).5. Tsao, K.W., deBarbieri, B., Hanspeter, M. & Waugh, D.W. A versatile plasmid expression vector for the production of biotinylated proteins by site-specific,enzymatic modification in Escherichia coli.Gene 169,59–64 (1996).6. Smith, P .A. et al. A plasmid expression system for quantitative in vivo biotinyla-tion of thioredoxin fusion proteins in Escherichia coli.Nucleic Acids Res.26,1414–1420 (1998).7. LaVallie, E.R. et al. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Bio/Technology 11,187–193(1993).8. Ong, E. et al. The cellulose-binding domains of cellulases: tools for biotechnolo-gy. Trends Biotechnol.7, 239–243 (1989).9. Smith, M.C., Furman, T.C., Ingolia, T.D. & Pidgeon, C. Chelating peptide-immobi-lized metal ion affinity chromatography. J. Biol. Chem.263, 7211–7215 (1988).10. Kim, J-S. & Raines, R.T. Ribonuclease S-peptide as a carrier in fusion proteins.Protein. Sci.2, 348–356 (1993).11. Su, X., Prestwood, A.K. & M cGraw, R.A. Production of recombinant porcinetumor necrosis factor alpha in a novel E. coli expression system.Biotechniques 13, 756–762 (1992).12. Nilsson, J., Ståhl, S., Lundeberg, J., Uhlén, M. & Nygren, P .Å. Affinity fusionstrategies for detection, purification, and immobilization of recombinant proteins.Protein Expr. Purif . 11, 1–16 (1997).13. Urry, D.W. Entropic elastic processes in protein mechanisms. I. Elastic structuredue to an inverse temperature transition due to internal chain dynamics. J.Protein. Chem.7, 1–34 (1988).14. Urry, D.W. Free energy transduction in polypeptides and proteins based oninverse temperature transitions. Prog. Biophys. Mol. Biol.57, 23–57 (1992).15. Urry, D.W. Physical chemistry of biological free energy transduction as demonstrat-ed by elastic protein-based polymers. J. Phys. Chem. B 101, 11007–11028 (1997).16. McPherson, D.T., Xu, J. & Urry, D.W. Product purification by reversible phase tran-sition following Escherichia coli expression of genes encoding up to 251 repeats of the elastomeric pentapeptide GVGVP . Protein Expr. Purif.7, 51–57 (1996).17. Hoffman. A.S. Applications of thermally-reversible polymers and hydrogels intherapeutics and diagnostics. J. Controlled Release 6, 297–305 (1987).18. Chen, J.P . & Hoffman A.S. Protein-polymer conjugates II. Affinity precipitationseparation of immunogammaglobulin by a poly(N -isopropylacrylamide)-protein A conjugate. Biomaterials 11, 631–634 (1990).19. Chilkoti, A., Chen, G-H., Stayton, P .S. & Hoffman, A.S. Site-specific conjugationof a temperature-sensitive polymer to a genetically-engineered protein.Bioconjugate Chem. 5, 504–507 (1994).20. Urry, D.W. et al. Temperature of polypeptide inverse temperature transitiondepends on mean residue hydrophobicity. J. Am. Chem. Soc.113,4346–4348(1991).21. Urry, D.W., Trapane, T.L. & Prasad, K.U. Phase-structure transitions of the elastinpolypentapeptide-water system within the framework of composition-tempera-ture studies. Biopolymers 24, 2345–2356 (1985).22. Vertesy, L., Oeding, V ., Bender, R., Zepf, K. & Nesemann, G. Tendamistat (HOE467), a tight-binding alpha-amylase inhibitor from Streptomyces tendae 4158.Eur. J. Biochem.141, 505–512 (1984).23. Porath, J. Immobilized metal ion affinity chromatography. Prot. Expr. Purif.3,262–282 (1992).24. Coligan, J.E., Dunn, B.M., Ploegh, H.L., Speicher, D.W. & Wingfield, P .T . Currentprotocols in protein science.(John Wiley & Sons, New York; 1995).25. Hartmeier, W. Immobilized biocatalysts.(Springer-Verlag, Berlin; 1988).26.Diamandis, E.P . & Christopoulos, T.K. Immun oassay.(Academic Press, SanDiego, CA; 1996).27. Dewhirst, M.W. & Samulski, T.V . Current Concepts: Hyperthermia in the treatmentof cancer. (The UpJohn Co., Kalamazoo, MI; 1998).28. Hauck, M.L., Dewhirst, M.W., Bigner, D.D. & Zalutsky, M.R. Local hyperthermiaimproves uptake of a chimeric monoclonal antibody in a subcutaneous xenograft model. Clin. Cancer Res.3, 63–70 (1997).29. Cope, D.A., Dewhirst, M .W., Friedman, H.S., Bigner, D.D. & Zalutsky, M .R.Enhanced delivery of a monoclonal antibody F(ab ′)2 fragment to subcutaneous human glioma xenografts using local hyperthermia. Cancer Res.50, 1803–1809(1990).30. Ausubel, F .M. et al. Current protocols in molecular biology (John Wiley & Sons,New York; 1995).31. Holmgren, A. & Bjornstedt, M. Enzymatic reduction-oxidation of protein disul-fides by thioredoxin. Methods Enzymol.107, 295–300 (1984).© 1999 N a t u r e A m e r i c a I n c . • h t t p ://b i o t e c h .n a t u r e .c o m。
Bacillus spp.

• Modification in milk proteins • Extraction of malt infusion • Clarification of beer • Production of flavouring derived from soy • Production of animal feeds
strains well characterized – Study of gene regulation of the enzymes of
interest
Enhancing
• Enhancing of strains – increasing production to tens of g/l in processes of 50- 100 hours lasting
Commodity chemicals
• Amilases & Proteases • Bacillus:
– Good catalytic properties – High cellular density from low cost carbon and
nitrogen sources – Realise directly to the culture medium (low cost
• enzymes for human food industry (beverage,dairy products, bakery foods)
• enzymes for animal feeds
50-60% of this market are enzymes from Bacillus American, European, Japanese, Chinese companies (Novozymes, Danisco-Genencor, DSM)
自噬与神经营养因子在运动干预阿尔茨海默病中的作用

阿尔茨海默病(Alzheimersdisease,AD)是一种 与年龄相关的不可逆的神经退行性疾病,其发病率 在世界范围内正逐渐增加。其临床表现主要为记忆 丢失和认知功能障碍,患者晚期独立生活能力丧失, 生活质量严重下降。AD的发病机制目前还未真正
的确定,但被广泛接受的机制为淀粉样蛋白级联假 说[1]。表现出细胞外 β淀粉样蛋白(betaamyloid, Aβ)的聚集物和神经细胞内神经原纤维缠结的病理 标志。目前临床上无法根治,治疗以减少 Aβ的产 生和加快 Aβ的清除为方向,以期减缓或延迟病情
收稿日期:20180213 修回日期:20180629 基金项目:陕西省自然科学项目(2014JM23027) 通信作者(correspondingauthor):liutaojd123@163.com
424
基础医学与临床 Basic& ClinicalMedicine
201939(3)
关键词:阿尔茨海默病;运动;自噬;神经营养因子
中图分类号:R74102 文献标志码:A
Roleofautophagyandneurotrophicfactors inexerciseinterventioninAlzheimersdisease
GUOShuqing,LIUTao
(DepartmentofHealthSciences,XianPhysicalEducationUniversity,Xian710068,China)
摘要:阿尔茨海默病(AD)是一种以渐进性记忆障碍为主要临床表现的神经退行性疾病。在临床药物无法根治的
现状下,运动干预带来的良好效应为治疗指引了新的方向。运动通过抑制 mTOR信号传导通路、刺激内质网应激、 加速线粒体损伤消除等自噬调节方式或加速 β淀粉样蛋白、tau蛋白的消除,或减弱它们的生成。同时,运动还对 神经元的凋亡起着调节作用。它提高了 BDNF、NGF等神经营养因子,激活了 PI3k/Akt途径,抑制了 caspase3和 COX2等凋亡因子的活性,从而减缓神经元的凋亡,减慢 AD的进程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Advances in protein solubilisation for two-dimensional electrophoresisTwo-dimensional (2-D)electrophoresis remains the highest resolution technique for protein separation and is the method of choice when complex samples need to be arrayed for characterisation,as in proteomics.However,in current proteome projects the total number of proteins identified from 2-D gels is often only a small percentage of the predicted proteome.In addition,there is an almost complete lack of hydrophobic proteins on 2-D gels,especially those using immobilised pH gradients.Recently there have been a number of publications reporting reagents which improve protein solubili-sation prior to isoelectric focusing.The improved solubilization possible with these reagents has increased the total number of proteins able to be visualised on 2-D gels and also allowed the separation of hydrophobic proteins,such as integral membrane proteins.Keywords:Two-dimensional polyacrylamide gel electrophoresis /Membrane proteins /Solubility /Proteome /ReviewEL 3428ReviewBen HerbertProteome Systems Ltd.,North Ryde,Sydney,AustraliaContents1Introduction ........................6602Chaotropes ........................6613Surfactants ........................6614Reducing agents ....................6625Sequential extraction .................6626Concluding remarks (6637)References ...... (663)1IntroductionAs we reflect on the third Siena meeting it is appropriate to look at what progress has been made in the various proteomic technologies towards achieving the goals of proteomics,for example,to identify and characterise all proteins expressed by an organism or tissue [1].The scope of this review is sample preparation for 2-DE and I would like to focus on the advances that relate to en-hanced protein solubility and increased numbers of hydro-phobic proteins on 2-D gels.For a comprehensive review of many other aspects of sample preparation for electro-phoresis,see Rabilloud [2].Two-dimensional electro-phoresis (2-DE)remains the highest resolution method for arraying proteins prior to their characterisation by mass spectrometry;however,the number of proteins ac-tually identified on 2-D gels,even from species where the entire genome is sequenced,is very low.Wilkins et al.[3]reported that for three species,Bacillus subtilis,Escheri-chia coli and Saccharomyces cerevisiae ,with completely sequenced genomes,less than 5%of the proteins in SWISS-PROT had been identified on 2-D gels.In addi-tion,within the 5%of identified proteins,only between 5%for S.cerevisiae and 15%for E.coli were hydrophobic,compared to the theoretical hydrophobic contents of 16%and 29%,respectively.An initial appraisal of these figures may lead to the conclusion that 2-DE and proteomics is failing to deliver;however,there have been a number of recent advances to sample preparation and 2-D gel meth-odology [4±7]which are already allowing more proteins to be arrayed in micropreparative quantities.Pretreatment of samples for isoelectric focusing (IEF)in-volves solubilisation,denaturation and reduction to completely break the interactions between the proteins and to remove nonprotein sample components such as nucleic acids [2].Ideally,to avoid protein losses,one would achieve complete sample solubilisation in a single step and thus eliminate unnecessary handling,as is the case for soluble protein samples which can be readily tak-en up in the most commonly used IEF sample solution of 8M urea,4%3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS),50±100m M dithiothreitolCorrespondence:Dr.Ben Herbert,Proteome Systems Ltd.,Locked Bag 2073,North Ryde,Sydney,NSW 1670,Australia E-mail:ben.herbert@ Fax:+61-2-9889-1805Abbreviations:ASB 14,amidosulfobetaine 14;GRAVY ,grand average of hydropathy;SB 3±10,N -decyl-N,N -dimethyl-3-ammo-nio-1-propane sulfonate;TBP ,tributyl phosphineWILEY-VCH Verlag GmbH,69451Weinheim,19990173-0835/99/0405-0660$17.50+.50/0(DTT)and40m M Tris.However,the`standard'IEF sam-ple solution is not ideal for many proteins and the chal-lenge for2-D PAGE,particularly in the context of proteo-mics,is the solubilisation and separation of insoluble samples such as membrane and membrane-associated proteins and proteins from highly resistant tissues like hair and skin.Enhanced protein solubility has been reported by introducing new reagents such as thiourea,sulfobe-taine surfactants and tributyl phosphine into the IEF sam-ple solution[4,5].To obtain a broad picture of a sample,the unfractionated proteins can be applied to a single2-D gel with a wide pH gradient,but with complex samples this approach only re-veals a small percentage of the anelle and plasma membrane fractions can be used to considerably reduce the complexity of cellular samples;however,they require considerable expertise and access to expensive equipment such as ultracentrifuges.In contrast,the sim-ple concept of sequentially extracting proteins by exploit-ing differential solubility has been used on many different tissues,usually with aqueous,organic solvent,and sur-factant-based extraction solutions[8±10].Recently Molloy et al.[6]have expanded on the sequential extraction ap-proach by incorporating highly solubilising conditions in the IEF sample solutions[4,5].Denaturing IEF sample solutions are made up of three main types of reagents, chaotropes,surfactants and reducing agents,and in the main part of this review I will summarise the advantages of some of the new reagents which constitute the en-hanced,highly solubilising IEF sample solution.2ChaotropesChaotropic agents such as urea allow proteins to unfold and thus expose their hydrophobic cores.This is a-chieved by changing the hydrogen bond structure in the solution,thus decreasing the energy penalty for contact of the hydrophobic residues with the solution[2,11].In practice,because the hydrophobic residues of proteins are exposed by urea denaturation it is normal to have sur-factants,such as CHAPS,present to aid in protein solubi-lisation.At the1996Siena meeting,and subsequently in a number of publications[4,12],T.Rabilloud has shown that proteins can be lost,by adsorption to the gel matrix, when IEF is conducted in immobilised pH gradients (IPGs).Rabilloud et al.[4]introduced the use of thiourea in combination with urea to increase the solubility of pro-teins in IPGs and showed that thiourea had a major posi-tive effect on the number of proteins visualised in the sec-ond dimension with four different subcellular fractions, including integral membrane proteins.Thiourea is an effi-cient chaotrope,although it is poorly soluble in water and requires high concentrations of urea for solubility,the opti-mal conditions being solutions of2M thiourea in5±7Murea.The enhanced protein solubility obtained by using thio-urea has also been demonstrated by Pasquali et al.[13],who showed improved2-DE of up to2mg of total mem-brane preparations and integral membrane preparationsfrom murine mammary epithelial cells.Fialka et al.[14] combined subcellular fractionation of murine mammaryepithelial cells with enhanced solubility using thiourea tocreate a range of organelle maps.They were able to sep-arate and identify some transmembrane proteins such asE-cadherin and caveolin.However,neither of these pro-teins would be classified as highly hydrophobic by thegrand average of hydropathy(GRAVY)measurementused by Wilkins et al.[3]to classify hydrophobic proteinsin B.subtilis,E.coli and S.cerevisiae.Although the useof thiourea/urea mixtures alone is a major advance in thesolubility of proteins for2-DE,the combination of these chaotrope mixtures with new sulfobetaine surfactants hasprovided a whole range of powerful sample solutions for2-DE.3SurfactantsAs stated in Section2it is normal to have at least one sur-factant present in the IEF sample solution to solubilise the hydrophobic residues that are exposed as a result of de-naturation in rge amounts of ionic substan-ces are not compatible with steady-state IEF;therefore,the use of SDS is not recommended and we are restrictedto nonionic or zwitterionic surfactants.It is possible to useSDS in the initial sample solubilisation and then dilute itout with a large excess of a nonionic surfactant[2];how-ever,obtaining sufficient dilution of the SDS can become impossible when micropreparative protein loads are re-quired.Traditionally,surfactants such as the Triton X-100and Nonidet P-40have been used,as well as sugar-based surfactants such as octyl glucoside[2,15].In re-cent years the sulfobetaine CHAPS has become the sur-factant of choice and is generally used at between2%and5%in8M urea.These commonly used surfactantsare soluble in high concentrations of urea,and thus itwould appear appropriate to add thiourea to the samplesolution and combine the enhanced chaotropic powerwith urea-soluble surfactants.However,while soluble,these surfactants are not efficient at protein solubilisationin high concentrations of chaotropes and their solubilisingpower is further minimised in the presence of highly cha-otropic thiourea[4,16].In contrast,sulfobetaines withlong linear alkyl tails such as N-decyl-N,N-dimethyl-3-am-monio-1-propane sulfonate(SB3±10)are more efficientthan CHAPS although they suffer from poor solubility inhigh concentrations of urea[17±19].Proteomicsand2-DEAs a compromise between chaotropic power and surfac-tant efficiency,Rabilloud et al.[4]suggested two IEF sample solution formulations which take advantage of the increased chaotropic power of thiourea.The first solution, 5M urea,2M thiourea,2%CHAPS and2%SB3±10is for proteins which require strong surfactants for solubility. The sulfobetaine SB3±10is not soluble in concentrations of urea greater than5M and thus the overall chaotrope concentration is relatively low.The second solution,7M urea,2M thiourea and4%CHAPS is for proteins which require a high concentration of chaotropes.To address the problem of surfactant-chaotrope compati-bility,Chevallet et al.[20]synthesised a range of novel sulfobetaines with more polar,but zwitterionic,head groups and long alkyl tails of more than12carbon atoms. The new surfactants were tested on membrane prepara-tions from bovine neutrophils and also on plasma mem-brane preparations from Arabidopsis thaliana.The most efficient surfactants were of the amidosulfobetaine type with either a linear alkyl tail,such as amidosulfobetaine 14(ASB14),or a mixed alkyl-aryl tail,such as C8F.The number refers to the number of carbon atoms in the tail. Because these surfactants have more polar head groups than sulfobetaines such as SB3±10they are soluble in high concentrations of urea,typically7±8M.When compared to the conventional IEF sample solution con-taining CHAPS,the new surfactants allowed the separa-tion and detection of many more membrane proteins. Specifically,C8F allowed the separation of some,quite hydrophobic proteins such as the PIP1isoform of a plas-ma membrane water channel protein from A.thaliana [20].PIP1has a GRAVY score of0.37,which rates it as one of the most hydrophobic proteins ever detected on2-D gels.In the case of the bovine neutrophil membranes the ASB14surfactant was highly efficient and resulted in many more proteins being visualised,including stomatin, a transmembrane protein[20].4Reducing agentsAs discussed above,reagents such as urea and surfac-tants are used to denature proteins and expose their hy-drophobic cores;however,to allow complete unfolding of many proteins it is necessary to reduce disulfide bonds. Reduction is usually achieved with a free-thiol-containing reducing agent such as b-mercaptoethanol or dithiothrei-tol(DTT)[2].However,free-thiol-containing reagents such as DTT are charged,especially at alkaline pH,and thus migrate out of the pH gradient during the IEF,which results in a loss of solubility for some proteins,especially those which are prone to interation by disulfide bonding, such as the keratins and keratin-associated proteins from hair and wool.Herbert et al.[5]replaced the thiol-contain-ing reducing agent DTT with an uncharged reducing agent,tributyl phosphine(TBP),which greatly enhanced protein solubility during the IEF and resulted in increased transfer to the second dimension with a range of samples including wool proteins.A further advantage of the mech-anism of phosphine reduction is that because phosphines do not contain a thiol they cannot be alkylated,which leads to a simplified IPG equilibration protocol incorporat-ing reduction and alkylation in a single ing an im-proved single-step IPG equilibration protocol[5],it is possible to combine TBP and an alkylating agent and ob-tain complete alkylation of cysteine.Because thiourea, sulfobetaine surfactants and TBP increase protein solubil-ity through different routes,they can be seen as comple-mentary reagents.Thiourea increases the chaotropic power of the sample solution,sulfobetaines such as SB 3±10,ASB14and C8F are efficient surfactants,and TBP ensures complete reducing conditions during the IEF; thus it is advantageous to combine all three in an en-hanced IEF sample solution.5Sequential extractionBy combining thiourea,sulfobetaines such as SB3±10 and the uncharged reducing agent TBP,it is possible to create a powerful IEF sample solution and thus solubilise proteins which would remain insoluble in conventional IEF sample solutions.However,the drawback of this en-hanced solubility is that for complex samples the2-D gels become swamped with overlapping proteins,especially when high loads are applied for micropreparative purpos-es.Molloy et al.[6]have adapted the concept of differen-tial solubility[8±10]by incorporating the enhanced solubi-lising conditions as the final step of a sequential extraction of E.coli.The first step is cell lysis and protein extraction using Tris base.The resulting pellet is extracted using conventional IEF sample solution,8M urea,4%CHAPS and DTT and the pellet remaining after this extraction is rich in membrane proteins and represents only11%w/w of the starting material.The final extraction is with5M urea,2M thiourea,2%CHAPS,2%SB3±10and2m M TBP.Eleven membrane proteins were identified on the 2-D gel from the final extraction,representing many of the outer membrane proteins of E.coli.Five of the identified proteins had not previously been identified from2-D gels and two of these were only previously known as open reading frames.It appears that many of the outer mem-brane proteins(OMPs)from E.coli were partitioned into the final pellet in a highly enriched state and were solubi-lised in the enhanced solubilising solution.Note that while the proteins in the final extract are clearly insoluble in conventional IEF solutions they are not rated as hydrophobic according to the GRAVY scale[3,6],asthey all have negative GRAVY scores.It seems that at the extremes of the GRAVY scale the values are predic-tive of protein solubility because the protein is mainly composed of either hydrophilic or hydrophobic residues. However,in the mid ranges of the GRAVY scale it is quite possible to have hydrophilic proteins,as measured by GRAVY,which are insoluble in the conventional IEF sam-ple solutions[6].The insolubility of a`hydrophilic'protein may be due to the presence of hydrophobic,possibly transmembrane,domains that are,on average,out-weighed by the majority of hydrophilic residues.Given the efficiency of some of the novel sulfobetaine sur-factants synthesised by T.Rabilloud et al.[20]it will be in-teresting to repeat the sequential extraction of Molloy et al.[6]using the new surfactants in the final step and look for more hydrophobic proteins.In another study,Molloy et al.[21]have used the novel sulfobetaine ASB14[20]as part of an enhanced solubilising solution to separate pro-teins extracted from E.coli with mixtures of chloroform/ methanol.The ASB14solution gave greater protein solu-bility,compared to a solution using SB3±10and CHAPS, probably because of the increased efficiency of the ASB 14and the fact that7M urea can be used with ASB14, compared with only5M urea with SB3±10.Out of the thir-teen proteins identified,eight had not been previously identified on2-D gels and five of these new proteins had positive GRAVY scores and are thus rated as hydropho-bic.6Concluding remarksThe past few years have seen2-DE undergo a revitalisa-tion which is strongly linked to the interest in proteomics. The high throughput nature of mass spectrometry,espe-cially MALDI-MS for peptide mass fingerprinting,has placed a large demand on2-DE to deliver large numbers of micropreparative gels of`unknown'proteins.It is clear that conventional technology,using urea,CHAPS,and DTT solubilisation on a single broad-range pH gradient is becoming exhausted.The same proteins are solubilised and identified many times,leaving behind the majority of the proteome,either insoluble or hidden behind the abun-dant proteins on a broad pH range2-D gel.The work summarised in this review has made significant progress on the protein solubility issue and has allowed many pre-viously insoluble proteins to be separated by2-DE.In ad-dition,the sequential extraction approach has proved to be a rapid and effective method of partitioning and con-centrating insoluble proteins such as membrane proteins.A combination of organelle fractionation with sequential extraction will prove to be very powerful.For example,se-quential extraction of purified organelles will provide cellu-lar localisation data as well as simplifying the2-D patterns by separating the soluble fraction from the membrane fraction.The next steps in the development of2-DE for proteomics will be continued refinements to prefractiona-tion and enhanced protein solubility,combined with2-DE using multiple pH gradients to cover the expected p I range of most proteomes.Received February17,19997References[1]Wilkins,M.R.,Sanchez,J.-C.,Gooley,A.A.,Appel,R.D.,Humphery-Smith,I.,Hochstrasser,D.F.,Williams,K.L., Biotechnol.Genet.Eng.Rev.1995,13,19±50.[2]Rabilloud,T.,Electrophoresis1996,17,813±829.[3]Wilkins,M.R.,Gasteiger,E.,Sanchez,J.-C.,Bairoch,A.,Hochstrasser,D.F.,Electrophoresis1998,19,1501±1505.[4]Rabilloud,T.,Adessi,C.,Giraudel,A.,Lunardi,J.,Electro-phoresis1997,18,307±316.[5]Herbert,B.R.,Molloy,M.P.,Walsh,B.J.,Gooley,A.A.,Bryson,W.G.,Williams,K.L.,Electrophoresis1998,19, 845±851.[6]Molloy,M.P.,Herbert,B.R.,Walsh,B.J.,Tyler,M.I.,Traini,M.,Sanchez,J.-C.,Hochstrasser,D.F.,Williams,K.L.,Gooley,A.A.,Electrophoresis1998,19,837±844. [7]Görg,A.,Obermaier,C.,Boguth,G.,Csordas,A.,Diaz,J.-J.,Madjar,J.-J.,Electrophoresis1997,18,328±337. [8]Weiss,W.,Postel,W.,Görg,A.,Electrophoresis1992,13,770±773.[9]Gurd,J.W.,Evans,W.H.,Perkins,H.R.,Biochem.J.1972,126,459±466.[10]Ramsby,M.L.,Makowski,G.S.,Khairallah,E.A.,Electro-phoresis1994,15,265±277.[11]Herskovitis,T.T.,Jaillet,H.,Gadegbeku,B.,J.Biol.Chem.1970,245,4544±4550.[12]Adessi,C.,Miege,C.,Albrieux,C.,Rabilloud,T.,Electro-phoresis1997,18,127±135.[13]Pasquali,C.,Fialka,I.,Huber,L.A.,Electrophoresis1997,18,2573±2581.[14]Fialka,I.,Pasquali,C.,Lottspeich,F.,Ahorn,H.,Huber,L.A.,Electrophoresis1997,18,2582±2590.[15]Righetti,P.G.,Isoelectric Focusing:Theory,Methodologyand Applications,Elsevier Biomedical Press,Amsterdam 1983,pp.1±386.[16]Navarette,R.,Serrano,R.,Biochim.Biophys.Acta1983,728,403±408.[17]Dunn,M.J.,Burghes,A.H.M.,Electrophoresis1983,4,97±116.[18]Gianazza,E.,Rabilloud,T.,Quaglia,L.,Caccia,P.,Astrua-Testori,S.,Osio,L.,Grazioli,G.,Righetti,P.G.,Anal.Bio-chem.1987,165,247±257.[19]Rabilloud,T.,Gianazza,E.,Catto,N.,Righetti,P.G.,Anal.Biochem.1990,185,94±102.[20]Chevallet,M.,Santoni,V.,Poinas,A.,Rouquie,D.,Fuchs,A.,Keiffer,S.,Rossignol,M.,Lunardi,J.,Garin,J.,Rabil-loud,T.,Electrophoresis1998,19,1901±1909.[21]Molloy,M.P.,Herbert,B.R.,Williams,K.L.,Gooley,A.A.,Electrophoresis1999,20,701±704.。