羟醛缩合
(完整)10.6羟醛缩合反应_

10-06 羟醛缩合反应
?
10-06 羟醛缩合反应
主讲人:肖咏梅
10-06 羟醛缩合反应
醛酮分子中,与羰基直连的a-碳原子的氢原子由于受羰基的影响使酸性增强
在稀碱的作用下含α-H的醛酮,能发生缩合,生成β-羟基醛或酮,称为羟醛缩合反应
α-H
机理: 共轭碱的负离子离域化,稳定性增强
ba
a,b不饱和醛
10-06 羟醛缩合反应
凡a碳上有氢原子的b-羟基醛都容易失去一分子水,生成a,b不饱和醛 具有a-H的酮也可发生类似反应,但一般产率很低
10-06 羟醛缩合反应
两种不同的带有a-H的醛、酮之间发生的羟醛缩合反应称为交叉的羟醛缩合反应
乙醛子间
丁醛分子间
a-C
a-C
乙醛的a-C进攻丁醛的羰基
丁醛的a-C进攻乙醛的羰基
10-06 羟醛缩合反应
发生羟醛缩合反应的条件

发生羟醛缩合反应的条件
羟醛缩合反应发生的条件主要包括:
1.存在羟醛化合物:羟醛缩合反应是由两个羟醛分子发生缩合反应而成的,因此必须要有羟醛化合物存在。
2.酸催化或碱催化:羟醛缩合反应可以通过酸或碱催化,使得反应速率得到加快,反应剂的物质量明显减少。
3.一定的温度和压力:羟醛缩合反应发生的温度一般在50℃以上,压力一般较低,一般在常压下即可。
4.适当的反应时间:羟醛缩合反应需要合适的反应时间,过长或过短都会影响反应的产率。
5.去除反应产物:随着反应的进行,羟醛缩合产物会逐渐生成,必须通过蒸馏、萃取等方法将其分离和去除,以促使反应向前推进。
羟醛缩合反应

O CH3CH
β β α
α
二、羟醛缩合反应的应用
应 用
增长碳链
有活泼的C=O、-OH
和C=C,易于转换成其
它化合物
三、羟醛缩合反应的类型
醛或酮的自身缩合 —— 相同的醛或酮之间
醛或酮的交叉缩合 —— 不同的醛或酮之间
四、提高羟醛缩合反应产率的实验措施
1. 自身缩合 醛
2. 交叉缩合
当两种不同的醛或酮都有α-H原子时,将会有 4 种羟醛缩合的产物。
分离困难,无实际应用价值!
醛
无α-H
醛 +
有α-H
酮
提供羰基
提供α -碳负离子
O HCH
CHO
O
CHO
①无 -H 的醛和有 -H 的酮缩合
CHO
+
O CH3CCH3
HO-
O CH=CHCCH3
苄叉丙酮
+
H2O
(产率78%)
交叉缩合
应用: 增长碳链,制备β-羟基醛(酮)或α,β-不饱和醛(酮) 及其它相关化合物 提高反应产率的实验措施: 改进反应装置or 改变反应物的加料方式 etc.
羟醛缩合反应
陈新
山梨酸
O OH-
2 CH3CHO
CH3CH=CH2CHO
CH3CCH3 Ba(OH)2 8H2O
O CH3CH=CH2CH=CHCCH3
羟醛缩合反应
一、羟醛缩合反应的概念
在稀碱催化下,含有α-H的醛或酮,生成α-碳负离子,进攻另一分子醛或酮
的羰基,发生亲核加成反应,生成β- 羟基醛、酮,或进一步脱水生成α,β-不饱
OH CH3 O
9.3.1 羟醛缩合反应
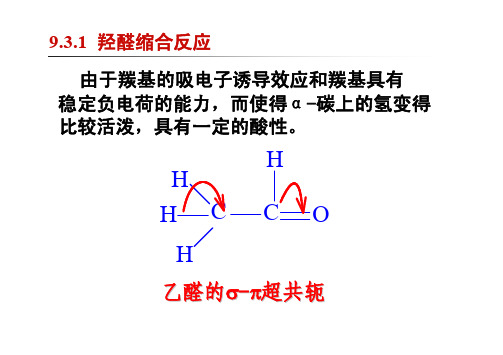
+-OH
+-OH -H2O
CH2CH3 C6H5 O
CH2CH3 O
缩合
C6H5
O
总结:
当分子中含有两个羰基且具有α-活泼氢时,总是优先 进行分子内的羟醛缩合,生成五元或六元环状不饱和 醛、酮。
CH3CH——CHCHO
OH H
CH3CH—CH2CHO O-
-H2O
CH3CH=CHCHO
9.3.1 羟醛缩合反应
+ CH3CH2CH2CH=O
H2CCHO CH2CH3
1MNaOH 80-100℃
பைடு நூலகம்
在较高的温度下得到 的是直接失水的产物
CH3CH2CH=C—CHO
+ H2O
CH2CH3
OH 70%
I2 -H2O
βα O
(CH3)2C=CHCCH3
=
=
9.3.1 羟醛缩合反应 ⑶ 分子内的缩合
O
O
(1) CH3CCH2CH2CH2CH2CCH3
KOH
CH3
O CH2CCH3
O
+-OH
CH3
β α C-CH3
O
加成失水
CH3
O CHCCH3
O
9.3.1 羟醛缩合反应
O
O
(2) CH3CH2CCH2CH2CH2CCH2C6H5
⑴ 醛的自身缩合
O
CH3CH + CH3CHO
稀NaOH 5℃
-H2O
室温
βα
CH3CH=CHCHO
α,β-不饱和醛
βα
CH3CHCHCHO OH H
β-羟基丁醛
2
9.3.1 羟醛缩合反应 反应历程:
羟醛缩合机理

羟醛缩合机理一、引言羟醛缩合是有机化学中一种重要的反应类型,也是生物大分子合成中最基本的反应之一。
本文将详细介绍羟醛缩合的机理。
二、羟醛缩合概述羟醛缩合是指在弱碱性条件下,甲醛或其他羰基化合物与具有活泼亲核性能的化合物(如胺、酚等)进行缩合反应,生成稳定的亚甲基或烷基化合物的过程。
在此过程中,甲醛分子首先被氢氧根离子(OH-)攻击,形成甲氧根离子和一个极性质子,然后极性质子进一步被亲核试剂(如胺、酚等)攻击,形成新的碳-碳键。
三、羟醛缩合机理1. 羟醛生成首先,在弱碱性条件下,甲醛分子与氢氧根离子发生反应,生成羟甲基或甲氧根离子。
这个步骤是整个反应过程中最重要的步骤之一。
2. 美拉德反应接着,在第一个步骤的基础上,羟甲基或甲氧根离子与亲核试剂(如胺、酚等)发生缩合反应,形成亚甲基或烷基化合物。
这个步骤被称为美拉德反应。
3. 羟醛缩合的条件羟醛缩合反应需要一定的条件,包括弱碱性环境、适当的温度和压力等。
在实际应用中,常用的催化剂包括氢氧化钠、氢氧化钾和三乙胺等。
四、羟醛缩合反应的机理解析1. 羰基亲核加成羰基亲核加成是指亲核试剂(如胺、酚等)中的负电子对攻击羰基碳上电子不足原子所形成的中间体。
在羟醛缩合反应中,亲核试剂(如胺、酚等)中的负电子对攻击了甲醛分子上电子不足原子所形成的中间体。
2. 产物稳定性分析产物稳定性是指产物分子内部键能与外界环境作用力之间平衡状态下所达到的最低自由能状态。
在羟醛缩合反应中,产物的稳定性是由产物分子内部键能和外界环境作用力之间的平衡状态所决定的。
3. 美拉德反应机理美拉德反应机理是指亲核试剂与甲醛分子发生缩合反应时,中间体的生成和消失过程。
在羟醛缩合反应中,美拉德反应机理包括亲核试剂攻击甲醛分子、中间体生成、中间体消失和产物生成等步骤。
五、羟醛缩合反应的影响因素1. 催化剂种类和用量催化剂种类和用量对羟醛缩合反应有重要影响。
常用的催化剂包括氢氧化钠、氢氧化钾和三乙胺等。
第三章:羟醛缩合

R''''
R' R'''
R' R'''
R' R'''
R' R'''
2,3-syn-3,4-syn
2,3-syn-3,4-anti 2,3-syn-3,4-anti 2,3-anti-3,4-anti
1.1 不对称控制方式
• 低物控制:非手性试剂(烯醇盐,烯丙基金属 试剂)对手性醛的加成。
• 试剂控制:手性亲核试剂对非手性醛的加成。手 性亲核试剂一般是通过酰化手性辅基或与手性配 体络合而产生手性特征。
Me
O
H Me
H
90% HO
Et Et
H
HO H
H
O
Me Me
H 10% HO
X
H
X
处理卤素和羟基时要慎重!!反常!! 量子化学计算
2.低物控制的羟醛缩合反应
2.1恶唑烷酮手性辅剂
O NH O
Ph O NH O
Evans酰胺手性硼烯醇盐易于制备,优异的立体选 择性,易于脱除和回收,倍收青睐。
OB(Bu'')2 R'CHO
SEt
syn-Si-Re
anti-Si-Si
-----------------------------------------------------------------------
手性二胺
T-(h)
Y%
syn:anti e.e%
-----------------------------------------------------------------------
羟醛缩合

使用铌酸(Nb2O5·nH2O)作为催化剂,进行丙酮气相羟醛缩合反应,发现反应产物种类与催化剂酸性中心 的酸强度和酸度密切相关。研究表明,铌酸催化剂表面的Brnsted酸性中心酸强度较强,在催化缩醛和缩酮的反 应中,具有很好的催化活性、选择性和稳定性。
羟醛缩合
化学反应
01 简介
目录
02 反应历程
03 反应机理
04 反应催化剂
05 有机化学中的应用
羟醛缩合,也叫做醇醛缩合,是指具有α-H的醛或酮,在酸或者碱催化下与另一分子的醛或酮进行亲核加成, 生成β-羟基醛或者β-羟基酮,β-羟基醛或者β-羟基酮可以受热脱水生成α,β-不饱和醛或酮。通过醇醛缩合, 可以在分子中形成新的碳碳键,并增长碳链。
碱金属化合物催化剂常用于羟醛缩合制备羟基醛的反应中,得到的产物经过加氢纯化可以得到二元醇乃至多 元醇,例如乙醛自缩合得到的3-羟基丁醛的反应,选择苛性钠水溶液作为催化剂,粗产品催化加氢可得到1,3-丁 二醇。同样的,甲醛和丁醛交叉缩合生成2,2-二羟甲基丁醛,选择碳酸钠和氢氧化钠混合溶液作为催化剂可以减 少副反应,提高反应选择性。
反应催化剂
酸性催化剂
碱性催化剂
常用的酸性催化剂有(VO)2P2O7、铌酸和MFI沸石等。在酸性催化剂的阳离子活性中心(Brnsted中心或 Lewis中心),醛羰基活化形成烯醇正碳离子从而发生缩合反应。酸催化的烯醇-酮平衡可表示为已有的研究发现, 催化剂表面酸性活性中心的种类、数目和分布都会影响其催化性能,适宜的酸强度能有效促进气相羟醛缩合反应 过程中碳正离子的形成,提高反应活性。
丙醛的羟醛缩合反应方程式

丙醛的羟醛缩合反应方程式丙醛是一种重要的有机化合物,常用于制药和农药工业中。
它是一种醛类化合物,分子式为CH3CHO,具有强烈的刺激性气味。
在化学反应中,丙醛可以发生羟醛缩合反应,生成2,4-二甲基-3-戊酮。
羟醛缩合反应是一种常见的有机反应,也称为肉桂酸合成反应。
该反应是一种加成-消除反应,通过将醛和羟醛反应形成α, β-不饱和羰基化合物来进行。
丙醛的羟醛缩合反应方程式如下所示:
CH3CHO + CH3OH → CH3CHOHCH(OMe)CH3
该反应需要催化剂存在,一般使用碱金属醇盐或稀酸催化剂。
反应条件通常在室温下进行,反应时间为几小时。
此时,丙醛和甲醇反应生成羟乙醛,然后再通过加成-消除反应生成2,4-二甲基-3-戊酮。
羟醛缩合反应可以合成许多不同的化合物,如肉桂酸、香兰素、黄酮类化合物等。
它是一种重要的有机合成反应,被广泛应用于制药和农药工业中。
在实际应用中,丙醛的羟醛缩合反应需要注意反应条件的选择和催化剂的选择。
此外,反应过程中需要保持反应体系的纯净和稳定,以确保产物的纯度和收率。
总之,丙醛的羟醛缩合反应是一种重要的有机合成反应,具有广泛的应用前景。
我们需要不断地深入研究和探索,在实践中不断改进反应条件和催化剂,为化学工业和生物医药行业做出更大的贡献。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第3章羟醛缩合和有关的反应3.1 引言开链化合物的立体控制反应在现代有机化学中是一个备受关注的问题,已发展出许多有用的方法用于具有刚性构象的复杂分子(如大环内酯合多环醛抗生素)的立体控制合成。
醛醇缩合反应在生物合成中是一种基本的键形成反应,因而受到特别的注意。
醛醇反应,即亲核试剂与亲电的羰基基团(及类似基团)的缩合反应,是构建不对称C-C键的最简单的,同时能满足不对称有机合成方法学的最严格要求的一类化学转化。
在复杂分子的合成和在光学活性的小分子砌块的制备中,可以找到许多不对称醛醇缩合反应的实例[1].在复杂的天然产物的合成中,常常会遇到制备具有多个相邻的手性中心的中间体的任务。
这类化合物的最有效的合成策略,是那种在连接二个反应片断的同时又建立起毗邻立体中心的策略。
在每一个前述的策略中,希望对于相对的(syn/anti)以及绝对的(R/S)立体化学都能实行控制 . 已有许多研究者报道了非对映选择性(对映选择性)醛醇缩合反应的结果。
这些不对称醛醇缩合反应中的主要变化因素是金属抗衡离子、与这些离子键合的配体以及反应条件。
下述几种方法可用于对醛醇反应进行不对称控制:105(1)底物控制:非手性烯醇盐或烯丙基金属试剂对手性醛的加成(一般在α-位).在这种情况下,按照Cram-Felkin-Ahn规则由优势过渡态来决定非对映选择性[2].(2)试剂控制:手性烯醇盐或烯丙基金属试剂对非手性醛的加成.最通用的获得手性烯醇盐的方法是通过手性辅剂以酯、酰胺(噁唑啉)、酰亚胺(噁唑烷酮)或硼烯醇盐的形式结合;手性烯丙基金属试剂通常也与手性配体结合.(3)双不对称反应:手性烯醇盐或烯丙基金属试剂对手性醛的加成.当醛和试剂显示互补的面优先性(匹配对的情况)时,能够提高立体选择性;反之当它们的面优先性相反(错配对)时,立体选择性降低.当与合适的配体配位时,许多金属抗衡离于(诸如Li,Mg,Zr,B,AI,Sb,Si,Ti)在不对称醛醇缩合反应中能提供良好的立体选择性.锂或镁形成络台物,它们通过Cram-Felkin-Ahn规则或配位控制加成提供选择性.钛的应用得到了极巧妙的、多样性的好结果,它与手性配体络合提供对映选择性的转化.硼烯醇盐由于其高对映选择性的传递性质而被证明具有广泛的用途.杂双金属催化剂和双核中心催化剂既能活化亲核试剂又能活化亲电试剂,它们贯穿了本章的讨论内容.可以说,只是从20世纪80年代早期开始,本论题才获得显著的进展.在本章中,我们试图介绍金属烯醇盐和有关的烯丙基金属衍生物对碳基化合物的加成反应的一些最重要的进展,如图3.1中的途径A和途径B所示的。
106通常,醛与金属烯醇盐的醛醇缩合反应在产物分子中产生出二个新的手性中心,导致四个可能的立体异构体2a,2b,2c和2d(图3.2,图3.3)107以硼参与的醛醇缩合反应为例,羰基化合物从垂直角度接近烯醇盐化合物,反应经历六元环过渡态.二烷基硼的烯醇式具有相对来讲较短的硼-配体键及硼氧键,这是有利于过渡态中1,3竖键的相互作用,也有利于连接醛中的羰基的基团在过渡态中采取假平键状态.因此,从椅型环状过渡态3a—3d(Zimmerman-Traxler模型[3],见图3.3),我们可以得出结论,烯醇盐的几何结构可以演变为产物的2,3-立体化学:Z-烯醇倾向于产生syn-产物(Z→syn),而E-烯醇主要产生anti-产物(E→anti);产物中3-羟基的绝对构型可由烯醇盐接近羰基的方向决定:即Re→syn,Si→anti.有几个参数对于立体化学控制是极为关键的:(1)烯醇盐中取代基部分的空间大小;(2)选择合适的试剂; (3)烯醇化作用所选用的条件.因此,Masamune[4]设计了二种类型的醛醇缩合试剂,它们在醛醇缩合反应中导致相反的立体化学.在下列结构中,化合物4可用于获得anti-醛醇缩合产物,而化合物5则可用来合成syn-醛醇缩合产物(图3.4)。
当醛醇缩合试剂5在n-Bu2BOTf和Et3N存在下与醛反应时,可以高度非对映选择性地产生syn-醛醇缩合产物6(表3.1).1083.2 底物控制的醛醇缩合反应3.2.1噁唑烷酮作为手性辅剂参与的醛醇缩合型反应1964年,Mitsui[5]首次使用手性辅剂获得了不对称醛醇缩合反应立体控制的结果,虽然那时的立体选择性还不高(58%).在20世纪80年代早期出现了显著的改进,当时Evans[6]和Masamune[7]引入了一系列导致高立体选择性的手性辅剂.在与二烷基硼烯醇盐结合时,这些手性辅基诱导了高对映选择性的醛醇缩合反应.109由N-酰基噁唑烷酮(如7和8)(在第2章它们被称为Evans辅剂并被用于羰基的(α-烷基化反应)生成的手性硼烯醇盐由于其易于制备、优异的立体选择性、易于脱除和回收使用而倍受青睐[8].通常,可通过将N-酰基噁唑烷酮与二正丁基硼三氟甲磺酸盐和三乙胺在CH2Cl2中于-78℃一起处理以制备Z-硼烯醇盐.这样得到的烯醇盐在同样的温度下容易发生醛醇反应,产生syn-醛醇缩合产物,非对映选择性大于99%(图3.5).在此,硼抗衡离子对立体选择性起着重要的作用.在烯醇化步骤中用三乙胺代替二异丙基乙基胺得到的结果更好.但是将硼换成锂则导致立体选择性下降.有人假设,立体选择性来源于二齿金属(如硼)通过椅型过渡态9与噁唑烷酮的羰基和烯醇氧配位的结果(图3.5)[9].110如图3.5所说明的,Evans酰胺7和8能以相反的立体化学的方式与醛进行醛醇缩合反应.应指出的是,这点和在第2章(2.4.3节)所述的分子内配位所得到的产物在羰基α-位的立体化学相反。
将酰胺10和12用于反应时,可得到一对对映体11和13(当11 中R=Me时)(图3.6)[6].111双不对称诱导也可应用于醛醇缩合反应.当手性醛15用含有非手性硼参与的烯醇盐14处理时,以1.75:1的比例得到非对映体混合物;然而,让同样的醛15与由Evans辅剂8衍生的烯醇盐反应时,以极高的立体选择性获得syn-醛醇缩合产物16(属于匹配对,观察到600:1的非对映选择性).将16转化为Prelog-Djerassi内酯17就只是一种常规的反应了.即使在错配对的情况下,例如用另一个Evans辅剂7处理醛15(就立体选择性而言,与8比较,7起相反的功用),仍能以极满意的非对映选择性获得产物18(400:1).当然,从这两个反应得到的最终产物互为非对映异构体(图3.7).化合物17,是所谓的(+)-Prelog-Djerassi内酯酸,从酒霉素(Methmycin)或幂霉素(narbomycin)降解而得,具有一系列大环内酯抗生素共有的重要的结构特征,并成为发展许多新的立体选择性合成的焦点.化合物17的制备示于图3.8[10].从8(R=Me)出发,用醛处理硼烯醇盐,通过不对称醛醇缩合反应在C-2和C-2’以预期的立体化学合成了化合物20;用亲电试剂处理8的锂烯醇盐在C-5以预期的立体化学生成19.请注意醛醇缩合反应和α-烷基化反应的立体化学彼此正好相反.将19和20偶合得到最终产物17。
通过醛醇缩合反应,下列α-乙烯基-β-羟基亚酰胺21’也用于天然产物的全合成.在所有21参与的醛醇缩合反应的例子中,都能得到98%以上的d.e.值(图3.9)[11]。
1121133.2.2 吡咯烷作为手性辅剂如上述,在各种天然产物如大环内酯、离子载体抗生素和其它乙酰型化合物(acetogenics)中经常出现β-羟基羰基单元,这刺激了这些化合物的立体控制合成方法学的发展,其中最为成功的方法是使用醛醇缩合反应[12].带有反-2,5-二取代的吡咯烷部分(作为胺组份)的酰胺烯醇盐22,已证明是不对称烷基化反应[13]和酰化反应[14]的优良底物.与成功的烷基化和酰化反应结果不同,在醛醇缩合反应中使用其锂烯醇盐不能得到好的立体选择性(表3.2,项1).另一方面,从相应的Li烯醇盐和二氯化二(环戊二烯)合法制得的Zr烯醇盐[15]则表现出明显的好的立体选择性(表3.2, 2~5项).114研究表明,带有大基团配体的锆原子专一地位于Z-烯醇盐平面的底部半球,醛分子从同侧接近并与Zr原子配位,形成椅型过渡态,导致赤式醛醇的形成(图3.10,图3.11).对于Li烯醇盐,在烷基化反应或酰化反应中烷基卤或酰卤的进攻则直接在烯醇盐的顶面发生.具有立体要求的金属中心在调节醛醇115反应中的重要性是显而易见的.从所确定的缩合产物的立体化学可知,这个反应的不对称诱导和先前讨论中看到的烷基化反应或酰化反应的立体化学结果是相反的.在脯氨酸型手性辅剂中也遇到类似的情况.Evans发现,由脯氨醇酰胺衍生的锂烯醇盐在烷基化反应中显示优异的非对映面选择性(第2章,2.4.2节,脯氨酸型手性辅剂),但在醛醇缩合反应中使用脯氨酸酰胺的锂烯醇盐却不成功.有效果的手性试剂是锆烯醇盐,它可以从相应的锂烯醇盐通过与Cp2ZrCl2的金属交换而得到.例如,在锆烯醇盐参与的醛醇缩合反应中,能够获得极佳的不对称诱导效果,syn/anti选择性达96%~98%,非对映面选择性达50~200:1(图3.12).116117酰化产物25可通过烯醇盐24与酰氯反应制得。
有趣的是,用Zn(BH4)2和KBEt3H处理酰化的烯醇盐25可分别得到syn-或anti-26(图3.13,表3.3)[16]。
虽然起始步骤是α-酰化反应,但最终产物仍可被看作醛醇缩合产物。
3.2.3 氨基醇作为手性辅剂以上所介绍得醛醇缩合反应都是在碱性条件下进行的,其中烯醇盐作为反应活性中间体参与反应。
在碱性条件下的醛醇缩合反应,其副产物有双聚体,高聚体,以及α,β-不饱和羰基化合物等。
与公认的碳阴离子化学相反,Mukaiyama发展了另一种实用的方法,其中烯醇是关键的中间体。
他首次证明用TiCl4和硅烷基醇醚作为稳定的烯醇等价物也能进行酸催化的醛醇缩合反应[17]。
继而,他又发展了硼三氟甲烷磺酸盐参与的醛醇缩合反应,此反应通过形成甲酰烯醇醚而完成。
Mukaiyama首次报道,硅烷氧基烯烃作为必需的潜在烯醇盐等价物在Lewis酸活化剂存在下与醛反应,这个方法现在称为Mukaiyama醛醇缩合反应(图3.14)。
在Lewis酸存在下,通过醛与硅烷基烯酮缩醛(在动力学控制下从丙酸酯生成)反应,在多数情况下可得到anti-醛醇缩合产物[18]。
在各种Lewis酸中,TiCl4几乎是最佳选择[17b]。
118在手性硅烷基烯酮缩醛的“Mukaiyama醛醇缩合反应”中,另一种用于控制绝对立体化学的手性辅剂由N-甲基麻黄碱衍生而得[26]。
它成功地应用于α-甲基-β-羟基酯(91%~94% e. e.)[19],[26]、α-甲基-β-羟基醛(91% e. e.)[20]、α-肼基和α-氨基酸(78%~91% e.e.)[21]、α-甲基-δ-氧代酯(72%~75% e. e.)[19b]、顺和反-β-内酰胺(70%~96% e. e.)[22]、carbapenem抗生素[23]和其它天然产物[24]的对映选择性合成。