价键理论
价键理论

主要的杂化类型和立体构型列于下表:
杂化类型
sp3
sp2
sp
sp3d或dsp3
sp3d2或d2sp3
立体构型
正四面体
正三角形
直线形
三角双锥体
正八面体
VSEPR模型
AY4
AY3
AY2
AY5
AY6
有d轨道参与的杂化轨道在配位化合物的价键理论中有介绍。
价Hale Waihona Puke 理论价键理论(Valence bond theory,VB理论)是一种获得薛定谔方程近似解的处理方法,又称为电子配对法。它是历史上最早发展起来的化学键理论。价键理论主要描述分子中的共价键及共价结合,核心思想是电子配对形成定域化学键。
其量子化学模型认为,共价键是由不同原子的电子云重叠形成的。例如,p电子和p电子可以有两种基本的成键方式:
价键理论中,为了解释分子或离子的立体结构,泡林以量子力学为基础提出了杂化轨道理论。其核心思想即是不同原子轨道的叠加重组,从而成为数目相同,能量相等的新轨道。例如,为了解释甲烷的正四面体结构,杂化轨道理论认为:
碳基态原子构型为1s22s22p2。首先碳2s中的一个电子被激发到空的2p轨道上,然后1个s轨道和3个p轨道重新组合成4个sp3杂化轨道,再分别和4个氢原子的1s电子成键。4个杂化轨道呈正四面体构型,键角109o28',能量没有任何差别。
1.电子云顺着原子核的连线重叠,得到轴对称的电子云图像,这种共价键叫做σ键。
2.电子云重叠后得到的电子云图像呈镜像对称,这种共价键叫做π键。
用形象的言语来描述,σ键是两个原子轨道“头碰头”重叠形成的;π键是两个原子轨道“肩并肩”重叠形成的。一般而言,如果原子之间只有1对电子,形成的共价键是单键,通常是σ键;如果原子间的共价键是双键,由一个σ键和一个π键组成;如果是叁键,则由一个σ键和两个π键组成。σ键可以是s-s,s-p,p-p等电子之间形成的,而π键可由p-p,d-p,d-d等电子之间形成的。除此之外,还存在十分多样的共价键类型,如苯环的p-p大π键,硫酸根的d-p大π键,硼烷中的多中心键,π酸配合物中的反馈键,Re2Cl82−中的δ键,等等。
高中化学—— 配合物的价键理论
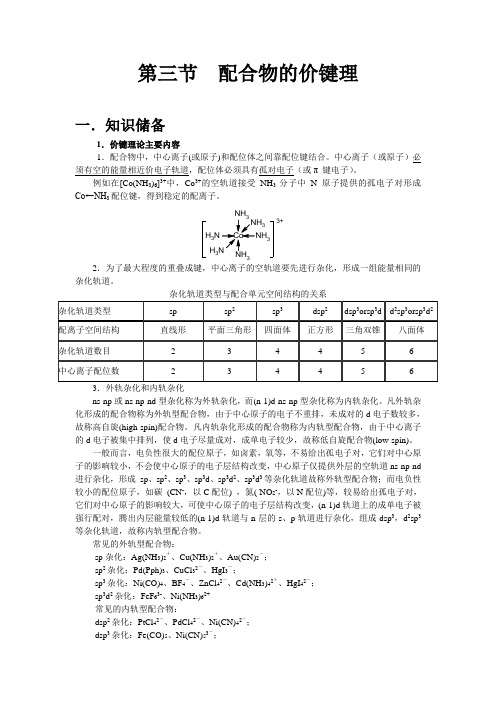
Fe3+ Fe2+ Co3+ Co2+ Mn2+ Fe3+ Co3+ Mn2+ Ni2+
sp3d2 sp3d2 sp3d2 sp3d2 sp3 d2sp3 d2sp3 d2sp3 dsp2
5
5.92
5.88 正八面体
4
4.90
4.为了增加配合物的稳定性,在某些配合物中除了形成 σ 配键外,还能形成反馈 π 配键。
(1)电中性原理:在形成一个稳定的分子或配离子时,其电子结构是竭力设法使每个 原子的静电荷基本上等于零。
(2)反馈 π 键:当配位体给出电子对与中心元素形成 σ 键时,如果中心元素的某些 d 轨道有孤电子对,而配位体有空的 π 分子轨道或空的 p 或 d 轨道,而两者的对称性又合适时, 则中心元素的孤对 d 电子也可以反过来给予配位体形成所谓的“反馈 π 键”。
1.A 的化学式 Cr(NH3)3O4 或 CrN3H9O4 A 的可能结构式如下图:
NH3
O
O
O
O Cr
O and/or O Cr
NH3
O
NH3
O
NH3
NH3
NH3
2.A 中铬的氧化数为+4 3.氧化还原性(或易分解或不稳定等) 4.化学方程式:CrO42-+3NH3+3H2O2=Cr(NH3)3(O2)2+O2+2H2O+2OH-
2.设配合物中碳原子数为 nC,则:nC︰nN=17.74/12︰31.04/14=0.667 已知 nN=2×2+2=6, 所以,nC=0.677×6=4 求出摩尔质量,由于剩余量过小,只能设 A 是氮氢化合物,由此得氢数,可
推得配体 A 为 H2NCH2CH2NH2,
H2C
配合物的结构示意图为:
Fe(CN)63-
化学竞赛—价键理论

π键的电子云
头碰头——σ键
肩并肩——π键
(1)π键的重叠程度不及σ键,键能较 小,键的活泼性较大,是化学反应 的参与者。 (2)所有共价单键均为σ键,共价双键 中有一个σ键和一个π键。 (3)普通σ键和π键为定域键,多个原 子间构成的大π键为离域键。
离域π键 由两个以上的 p轨道以“肩并肩” 的方式重叠形成的键,称为离域π键或 大π键。 n 通常离域π键用符号π m 表示,n表 示p轨道数,也是成键的原子数,m表 示电子数。
二、 价键理论
离子键理论能很好地说明离子化合物的形 成和性质,但不能说明由相同原子组成的单质 分子(如H2、Cl2、N2等),也不能说明不同非金 属元素结合生成的分子,如HCl、CO2、NH3等 和大量的有机化合物分子形成的化学键本质。 1916年美国化学家路易斯(G N Lewis, 1875~1946)提出了共价学说,建立了经典的共 价键理论。原子间可共用一对或几对电子,以 形成稳定的分子。
N N
键长:成键两原子间的平均间距。
在101.3kPa,298K条件下,断开1molAB(g)分子 中的化学键,使其分别变成气态A原子和气态B原子 所吸收的能量。
共价键 键长 键能 l/pm E/(kJ· -1) mol 92 570 H-F 432 H-Cl 127 366 H-Br 141 161 298 H-I 141 159 F-F 243 Cl-Cl 198 193 Br-Br 228 267 151 I-I 共价键 键长 键能 l/pm E/(kJ· -1) mol 436 H-H 74 346 C-C 154 C C 134 602 C C 120 835 159 N-N 145 N N 110 946 414 C-H 109 464 O-H 96
化学竞赛-价键理论

价键理论强调了电子在形成化学 键中的重要作用,并解释了分子 中的键合类型、键合强度和分子 几何结构等方面的信息。
价键理论的发展历程
19世纪初,价键理论的基本概念开始萌芽,当时科学 家们开始认识到电子在化学键中的作用。
20世纪初,价键理论得到了进一步的发展和完善,其 中最为著名的科学家是英国化学家莫里斯·威廉·皮尔兹
洪特规则
洪特规则是描述电子排布和化学键形成的规则之一,它指 出在相同能级的不同轨道中,电子优先以自旋方向相同的 方式占据轨道。
在形成化学键时,洪特规则可以预测电子在轨道中的排布 方式,从而影响化学键的类型和稳定性。
洪特规则可以解释许多化合物的电子构型和几何构型,是 理解和预测化学反应的重要工具。
03 价键理论的实际应用
分子的几何构型
分子构型的确定
价键理论不仅解释了共价键的形成,还为确定分子几何构型提供了理论基础。根 据电子对的排斥作用和最小化能量原则,可以预测分子的几何构型。
分子构型的稳定性
分子的几何构型不仅决定了分子的物理和化学性质,还影响了分子的稳定性。根 据价键理论,电子对的最大重叠和最小排斥原则有助于理解分子构型的稳定性。
以及金属与配体的相互作用。
无机化学
价键理论可以用来描述无机化合物的分子 结构和性质,如氢化物、氧化物和含金属 的化合物等。
生物化学
虽然价键理论在生物化学中的应用相对较 少,但它仍然可以用来描述某些生物分子 的结构和性质,如蛋白质和核酸等。
02 价键理论的基本原理
电子配对原理
01
电子配对原理是价键理论的核心,它指出原子在形成化学键时, 倾向于将未成对的电子配对成键,以使系统能量最低。
共价键的形成与断裂
共价键的形成
价键理论

+
C
C
成键
C
C
π
HA + HB H2 φ1 - φ2 + HA φ1 HB φ2 成键 ψ1=φ1 + φ2 反键 ψ2=φ1 - φ2 φ1 φ2 φ1 + φ2 ψ1 ψ2
电子在分子轨道中的填充顺序
遵循
能量最低原理 泡里不相容原理 洪特规则 最大重叠 能量相近 对称性匹配
π*
此外还遵循成键三原则:
节面 C C 反键 C C
共价键
既难失去四个 电子C+4 也难得到四个 电子 C-4
C,第二周期,第四主族元素
与其它的原子共享四对电子, 达到外层8电子的稳定结构。
共价键
有机物与无机物的显著差别是由其组成和结构 ห้องสมุดไป่ตู้定的
有机物 无机物 O ①. 由C、O组成 2 CO2+H2O+Q (易燃) 共价键 离子键 ②. 分子间作用力~范德华力m.p.低 离子晶格—高m.p. 相似相溶 易溶于极性的水 ③. 难溶于极性的水 ④. 分子间碰撞——反应, 极快,故产率高 不是所有的碰撞都能反应 活化能高碰撞的位置、方向合适 反应部位多,副反应多
一、价键理论
两原子都有未成对电子 且自旋相反 原子轨道重叠 (电子匹配) 成键(定域键)
共价键的 最大重叠原理: 饱和性: 方向性: 例 H2 + Cl2 y + H(1S) x
2HCl
y x 重叠最大 x 部分重叠 x
二、分子轨道原理
从分子的整体出发研究分子中每个电子的运动状态 形成化学键——电子在整个分子中的运动(离域键) 分子轨道——由能量相近的原子轨道线性组合而成。
价键理论

于是, CO 可表示成 :
C
O
配位键形成条件: 一个原子中有对电子;而另一原子中有可 与对电子所在轨道相互重叠的空轨道。 在配位化合物中,经常见到配位键。 在形成共价键时,单电子也可以由对电子分开而得到。如 CH4 分子, C 原子 2s2 2p2 ,只有 2 个单电子。 2s 2p 2s 2p
2 价键理论 将对 H2 的处理结果推广到其它分子中,形成了以量子力学 为基础的价键理论 ( V. B. 法 ) 。 1°共价键的形成 A、B 两原子各有一个成单电子,当 A、B 相互接近时,若 两个电子所在的原子轨道能量相近,对称性相同,则可以相互重 叠,两电子以自旋相反的方式结成电子对。于是体系能量降低, 形成化学键。一对电子形成一个共价键。 形成的共价键越多,则体系能量越低,形成的分子越稳定。 因此,各原子中的未成对电子尽可能多地形成共价键 。 例如,H2 中,可形成一个共价键; HCl 分子中,也形成一个 共价键。 N2 分子怎样呢 ?
这说明破坏 H2 的化学键要吸热 ( 吸收能量 ),此热量 D 的 大小与 H2 分子中的键能有关。 计算还表明,若两个 1s 电 子以相同自旋的方式靠近,则 r 越小,V 越大。此时,不形 成化学键。 如图中上方红色曲 线所示,能量不降低 。
0 V
-D
ro
r
H2 中的化学键,可以认为是电子自旋相反成对,结果使体 系的能量降低 。 从电子云的观点考虑,可认为 H 的 1s 轨道在两核间重叠,使电子在两核间出现 的几率大,形成负电区。两核吸引核间负 电区,使 2 个 H 结合在一起。
3s
3p
激发
3d
3s 3pFra bibliotek3d 激发后,有 5 个单电子,与 5 个 Cl 形成共价键 。
大工普通化学第一章
H2分子形成过程能量随核间距变化示意图 E
☺
排斥态 0
基态
0
74pm
R
共价键的特点:
•饱和性 H Cl H O H •方向性
1. 价键理论(valence bond theory)
共价键( covalend bond):原子间通过共 用电子对形成的化学键。
价键理论基本要点: •相邻两原子间只有自旋方向相反的两个电子 相互配对时,才能形成稳定的化学键。 •原子轨道重叠时,只有同号轨道能发生有效 重叠,且重叠越多成键越牢固。
sp2杂化 B: 2s22p1
2p
2s
BF3的空间构型 为平面三角形
F
B
F
F
2s
2p 激发 2s 2p
sp2 sp2杂化
BF3的形成
三个sp2杂化轨道
sp3杂化 C:2s22p2
2p
2s
CH4的空间构 型为正四面体
2s
2p 激发 2s 2p
sp3杂化
sp3
CH
的形成
4
四个sp3杂化轨道
不等性sp3杂化 NH 3 N: 2s22p3
原子中不同类型原子轨道混合,重新组成 新的原子轨道的过程称为原子轨道杂化,新的 原子轨道称为杂化轨道。
基本要点: •成键时能级相近的价电子轨道混合杂化, 形成新的价电子轨道——杂化轨道(hybrid orbital)。 •杂化前后轨道数目不变。 •杂化后轨道伸展方向,形状发生改变。
杂化轨道的类型有: ns与np的杂化, 如:sp, sp2, sp3等性杂化, sp3
价键理论
1、价键理论:以原子轨道作为近似基函数描述分子中电子的运动规律,在阐述共价键本质时,根据Pauli原理的要求,认为一对自旋反平行的电子相互接近时,彼此呈现互相吸引的作用,使体系能量降低,形成化学键。
2、价键理论和分子理论的比较:(1)在数学处理上选用的变分函数不同价键法以原子轨道作为基函数,进行变分法处理,定变分参数;MO(分子理论)法中,每个分子轨道都涉及整个分子,具有离域键概念。
(2)由于选用的基函数不同,所得结果也不相同(3)VB法和MO法在其初级价段都是粗略的近似方法,各有其优缺点,而改进后,两者的结果就彼此接近了。
(4)两者的电子云都在核间密集。
在MO法中,把电子云过多的几种到核间,引起排斥能增大,算得的E偏高,因而求得H2分子的解离能偏低了。
(5)将VB法和MO法推广应用到其他多原子分子:VB法用定域轨道概念描述分子的结构,配合杂化轨道法,适合于处理基态分子的性质;MO法中每个分子轨道都遍及整个分子整体,而分子中各个分子轨道都具有一定的分布和能级,非常适合于描述分子的基态和激发态的性质。
3、价电子对互斥理论:原子周围各个价电子对之间由于相互排斥,在键长一定的条件下,互相间距离愈远愈稳定。
这就要求分布在中心原子周围的价电子对尽可能离得远些,由此说明许多简单分子的几何构型。
4、杂化轨道理论:原子在化合成分子的过程中,根据原子的成键要求,在周围原子影响下,将原有的原子轨道进一步线性组合成新的原子轨道。
这种在一个原子中不同原子轨道的线性组合,称为原子轨道的杂化。
杂化时,轨道的数目不变,轨道在空间的分布方向和分布情况发生改变,能级改变。
组合所得的杂化轨道一般均和其他原子形成较强的键或安排孤对电子,而不会以空的杂化轨道的形式存在。
5、离域分子轨道理论:用分子轨道理论处理多原子分子时,最一般的方法是用非杂化的原子轨道进行线性组合,构成分子轨道,它们是离域化的,即这些分子轨道中的电子并不定域在多原子分子中的两个原子之间,而是在几个原子间离域远动。
价键理论
价键理论价键理论valence-bond theory,一种获得分子薛定谔方程近似解的处理方法。
又称电子配对法。
历史上最早发展起来的化学键理论。
主要描述分子中的共价键和共价结合,其核心思想是电子配对形成定域化学键。
1产生1927年W.H.海特勒和F.W.伦敦首次完成了氢分子中电子对键的量子力学近似处理,这是近代价键理论的基础。
L.C.鲍林等加以发展,引入杂化轨道概念,综合成价键理论,成功地应用于双原子分子和多原子分子的结构。
价键理论与化学家所熟悉的经典电子对键概念相吻合,一出现就得到迅速发展。
但价键理论计算比较复杂,使得后来发展缓慢。
随着计算技术日益提高,该理论还会有新发展。
1927年,Heitler 和London 用量子力学处理氢气分子H2,解决了两个氢原子之间化学键的本质问题,使共价键理论从典型的Lewis理论发展到今天的现代共价键理论。
海特勒-伦敦方法处理氢分子氢分子的哈密顿算符是:式中rA1、rB1为核A、B与电子1之间的距离;r12为两个电子之间的距离;RAB为两个原子核之间的距离……(图1);1/RAB表示两个原子核之间的势能(氢核和电子电荷皆为1基本电荷单位);1/rA1、1/rB1、…也是势能;墷是拉普拉斯算符。
海特勒-伦敦方法的要点在于如何恰当地选取基态H2的近似波函数Ψ(1,2)(或称尝试波函数),然后用变分公式使氢分子能量E为最低(假定Ψ是归一化的):式中*表示复数共轭。
考虑两个氢原子组成的体系,若两个氢原子A(有电子1)和B(有电子2)的基态波函数为:φA⑴=πexp(-rA1)φB⑵=πexp(-rB2)假如两个氢原子相距很远,那么体系波函数是:Φ1(1,2)=φA⑴φB⑵实际上两个电子是不可区分的。
同样合适的函数是:Φ2(1,2)=φB⑴φA⑵两个函数Φ1和Φ2都对应相同的能量。
海特勒和伦敦就取两个函数的等权线性组合作为H2的变分函数:Ψ(1,2)=c1Φ1+c2Φ2解久期方程得c1=±c2,波函数和能量是:式中s称原子轨道的重叠积分。
价键理论
2杂化轨道类型
(1)SP杂化:每个轨道中S与P各占 二分之一,头大尾小(s成分大),直 线型。如:BeCl2 。Be2+(1s22s2)
2) SP2 杂化
,每个轨道中S成分为1/3,P为2/3,头 变小,尾变大,正三角形。如 BF3 。
3)SP3 杂化
每个轨道中S成分为1/4,头更变小。为 正四面体,如CCl4 。
二.杂化轨道理论
hybrid orbital 1要点: 1) 原子在形成分子时,为增强成键能力, 不同类型的原子轨道(必要时电子可激 发到能量相近轨道)可以重新组合成新 轨道,叫杂化轨道。 2) 杂化轨道数=参与杂化的原子轨道数 3) 参与杂化的轨道要能级相近 4) 不同类型的杂化轨道的形状与空间取 向不同。
一. 经典价键理论
classical valence-bond 1 要点: 1. 具有成单电子的两原子轨道相互重叠, 可以形成共价键。重叠越大,键越稳定 2. 共价键具有方向性和饱和性。(原因) 3. 根据重叠方式不同,可分为σ键和π 键。头碰头式与肩并肩式。 π键键能较 σ键小。(如N2)
杂化轨道理论仍保存着经 典理论的三大要求
3.等性杂化与不等性杂化
全部由成单电子的轨道参与的杂化叫等 性杂化。有孤对电子的轨道参与的杂化 叫不等性杂化。 NH3 分子的形成,有一个孤对电子参与 杂化,键角变小。分子构型:三角锥。
sp3杂化
2p
2s
sp3杂化轨道
H2O 分子的形成,有两个孤对电子参与杂化, 键角变得更小。分子构型:V型。
三.价层电子对互斥理论
这种理论常能更方便地推断出分子的空间构型。 1理论要点: 1) 先确定价层电子对数。在此公式中,O和S 原子作为配体时,接受其它原子的配位 2) 根据价层电子对数确定其空间构型。 3) 价层电子对数=中心原子的配位数时,价层 电子对的构型与分子的构型相同。否则不同。 4)根据孤对电子数来推断分子的空间构型。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
价键理论
自1916年路易斯提出经典的共价键理论以来,共价键理论有了很大的发展。
现代共价键理论有两种,一是价键理论,二是分子轨道理论。
(一)价键理论的基本要点
价键理论,又称电子配对法,其基本要点如下:
1.具有自旋相反的未成对电子的两个原子相互接近,可以形成稳定的共价键。
如果A、B两个原子各有一个自旋相反的未成对的电子,那么这两个未成对电子可以相互配对形成稳定的共价键,这对电子为A、B两原子所共有(共用)。
如果A、B各有两个或三个未成对的电子,则自旋相反的单电子可两两配对形成双键或叁键。
如果A原子有两个未成对电子,B原子有一个未成对电子,那么一个A原子能与两个B原子结合形成AB2型分子,…。
2.原子中未成对的电子数等于原子所能形成的共价键数目(共价键的饱和性)。
共价键是由成键原子中自旋相反的未成对电子配对形成的。
一个原子的一个电子和另一个原子的一个电子配对以后,不能再和第二个电子配对。
因为这时其中必有两个电子的自旋方向相同而相斥。
也就是说一个原子所能形成共价键的数目是一定的。
原子中未成对的电子数等于原子所能形成的共价键数目,这就是共键价的饱和性。
例如,H原子只有一个未成对电子,它和另一个H原子的未成对电子配对后,就不能再与第二个H原子的电子配对了,……。
3.成键电子的电子云重叠越多,核间电子子云密度就越大,形成的共价键就越牢固(共价健的方向性)。
共价键的生成是由于自旋相反的单电子相互配对,电子云重叠的结果。
因此,当两个原子形成分子时,电子云重叠的程度越大,则两原子间的电子云密度越大,生成的共价键就越牢固,所以,在形成共价键时,电子云总是尽可能达到最大程度的重叠。
因此,在形成共价键时,原子间总是尽可能沿着电子云最大重叠方向成键。
s电子云呈球形对称分布,p、d、f电子云在空间都有一定的伸展方向。
在形成共价键时,除了s 电子云和s电子云可以在任何方向上都能达到最大程度的重叠外,p、d电子云的重叠,只有在一定方向上才能使电子云有最大程度的重叠。
即共价键是有方向性的。
例如,当氢原子1s电子云和氯原子的3p电子云重叠形成HCL分子时,氢原子的1s电子云总是沿着氯原子未成对电子的3p电子云对称轴方向作最大程度的重叠(图4-9(a))。
其他方向都不能形成稳定的分子(图4-9(b)(c))。
电子云的三种重叠情况
图4-9 氢原子的1s电子云与氧原子的3P
x
(二)共价键的类型
共价键有两种成键方式。
一种是电子云以:“头碰头”方式相重叠,电子云及重叠部分沿键轴(两核间连线)呈圆柱形对称分布,重叠部分绕轴旋转任何角度形状不会改变,这种键叫σ键。
另一种是成键的两个电子云的对称轴相平行,以“肩并肩”方式相重叠,电子云重叠部分对通过键轴的一个平面具有对称性,这种键称为π键。
例如在N2分子中,氮原子的价层电子结构为:2p x12p y12p z1三个未成对的p电子分占三个互相垂直的p轨道。
当两个氮原子结合成N2分子时,p x电子云沿x轴方向以“头碰头”方式重叠形成一个σ键,每个原
子剩下的两个p电子云不能再沿x轴方向“头碰头”重叠,只能让p电子云的对称轴平行,以“肩并肩”方式重叠形成两个π键。
如图4-10。
4-10 N2分子形成示意图
由于σ键电子云重叠程度较π键大,因而σ键比π键牢固。
一般来说,π键容易断开,化学活泼性较强。
π键不能单独存在,只能与σ键共存于具有双键或叁键的分子中。
σ键不易断开,是构成分子的骨架,可单独存在于两原子间。
以共价键结合的两原子间只能有一个σ键。