电子转移步骤动力学完整版
电化学理论与方法 第七章 电子转移动力学

j 0 越大,净反应速度也越大,电极反应越 当极化值相等时,
容易进行。
以同一净反应速度进行,则 j 0 越大,其可逆性越大 。
0 越大,所需要的极化值越小。 j
通过交换电流密度的大小,有助于判断电极反应的可逆性。
电极反应的可逆性:电极反应这种力图恢复平衡态的能力,或 者说去极化作用的能力称之为~。
交换电流密度值与电极体系动力学性质之间的关系
§6.2.3电极反应速度常数K
1、定义 电极电位为标准电极电位和反应粒子浓度为单位浓度时电 极反应的绝对速度,单位为cm/s,它本身排除了浓度变化的 影响。 K的表达式: F 0 F 0 F Kc R exp F KcO exp RT RT 已知
第6章 电子转移步骤动力学
内容
电极电位对电子转移步骤反应速度的影响 电子转移步骤的基本动力学参数 稳态电化学极化规律 多电子的电极反应 双电层结构对电化学反应速度的影响 电子转移步骤量子理论简介
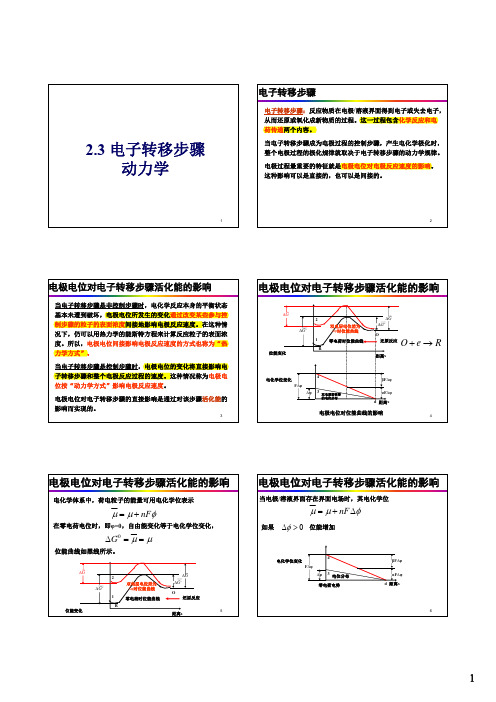
§6.1.1电极电位对电子转移步骤活化能的影响
电极电位对电子转移步骤的直接影响是通过对该 步骤活化能的影响实现的。 反应活化能:反应物越过反应能垒而活化所需的 能量。
(6.8)
式中 即
K 和 K 分别表示电位 0 处的反应速度常数,
用 j 和 j 分别表示电位 0 处的还原反应速度和氧 化反应速度,则
0
0
j F Kc O j F KcR
代入式(6.8)、式(6.9)得
0
0
(6.12)
(6.13)
F j j exp RT
F 平 F 平 F Kc R exp j F KcO exp RT RT
第四讲电子转移步骤动力学
电极电位对电子转移步骤活化能的影响电极电位对电子转移步骤活化能的影响电极电位对电子转移步骤活化能的影响
电极电位对电子转移步骤活化能的影响
电极电位对电子转移步骤活化能的影响
G
∆
0G F αϕ∆+
同一个电极上发生的还原反应和氧化反应的绝对速度与电极电位交换电流密度交换电流密度
交换电流密度交换电流密度
交换电流密度交换电流密度
交换电流密度稳态电化学极化规律稳态电化学极化规律稳态电化学极化规律
稳态电化学极化规律
η
高过电位下的电化学极化规律
2.3 2.3
高过电位下的电化学极化规律
电极反应以一定的速度进行时,电化学过电位的大小取决于电极
混合控制时的动力学规律已知混合控制时的动力学规律
i i
RT RT
i i
RT RT i i
RT RT
i i RT RT。
第六章电子转移步骤动力学

第六章电子转移步骤动力学电子转移是指电子从一个原子或分子向另一个原子或分子的跃迁。
这个转移过程可以通过多种方式发生,比如离子交换、电子传递等。
在化学反应中,电子转移步骤是非常重要的,对反应速率和产物选择性有着显著影响。
因此,研究电子转移步骤动力学是理解和控制化学反应的关键。
电子转移过程中,电子的跃迁通常伴随着原子或分子的几何变化。
这种几何变化可以通过分子轨道理论和能带理论等方法进行描述。
通过这些理论,我们可以计算得到电子转移过程的能垒和反应路径,并进一步分析其动力学特性。
在电子转移步骤的动力学研究中,我们经常使用催化剂来促进转移过程。
催化剂可以提供一个较低的能垒,从而加速反应速率。
催化剂通常通过与反应物形成配位键或中间体来实现对反应的加速作用。
通过研究催化剂的结构和反应机理,我们可以深入理解电子转移步骤的动力学行为。
另一个重要的研究方法是通过动力学实验来研究电子转移步骤。
动力学实验的基本原理是测量反应速率随时间的变化。
通过改变反应条件,如温度、压力和浓度等,我们可以探究电子转移步骤的温度和压力依赖性,以及反应物浓度对反应速率的影响。
通过这些实验数据,我们可以确定反应的速率常数和反应级数,并进一步分析反应机理。
除了实验研究,理论计算也在电子转移步骤动力学研究中发挥了重要作用。
量子力学和分子动力学模拟等计算方法可以提供详细的分子层面的信息,如反应势能面、反应路径和能量障碍等。
通过理论计算,我们可以预测反应速率常数、反应物选择性和反应动力学行为,为实验研究提供重要参考。
总之,电子转移步骤动力学研究是理解和控制化学反应的重要手段。
通过研究电子转移过程的动力学特性,可以揭示反应机理和反应条件对反应速率和产物选择性的影响。
这一领域的研究对于发展新型催化剂、设计高效反应体系以及理解生命现象等具有重要意义。
第三章4电子转移步骤动力学

)]
可以简化为
ic
i0[1
anFc
RT
(1
nFc
RT
)]
i0
nFc
RT
与经验公式的比较
对比公式 i 可得:
RT 1
nF i 0
定义极化电阻:
Rr
d
di
0
RT nFi 0
五. 弱极化区的极化规律
在这个极化区域内,B-V方程不可简化,必 须用完整得B-V公式描述动力学规律:
K k exp G0
RT
令: i0 nF KcO 则:
i 0 nF KcR
i i0 exp nF
RT
i i0 exp nF
RT
将上式取对数整理后:
2.3RT log i0 2.3RT log i
O*
能变化;
RR,:Ag 自晶格中
逸出的位能变化; G 0
RAO :Ag 在相间
转移的位能曲线; R
R*
A G0
O
dx
界面电场对活化能的影响
G G0 nF G
G 0
G G0 nF
1
传递系数
F
G0 G
d
x
F
ic=i
i
i0
exp
nF
RT
c
exp
nF
RT
c
六. 用稳态极化曲线法测量动力学参数
c
log j
0
log j
log jc
电子转移步骤的动力学

G G0 nF, G G0 nF
jc
nF zc
exp
G 0
nF
RT
c0O
jc0
exp
nF
RT
ja
nF za
exp
G0 nF
RT
c0R
ja0
exp
nF
RT
上式即为电极电位对电极反应绝对速度影响 的公式,也即电子交换步骤的基本方程式。
注:1.上述公式中电位零点的选取是任意的,无 具体物理含义;
2.绝对电流密度是无法直接测量到的,又称 内电流密度,所能测量的是内电流密度的差值, 净电流密度。
将绝对电流密度表达式写成对数形式有:
2.3RT
nF
lg
ja0
2.3RT
nF
lg
ja
2.3RT
nF
lg
jc0
2.3RT
G 0
nF (
平)(还原)
G
G 0
nF
(
平)(氧化)
G0,G0分别为 平时的还原与氧化
过程的活化能
其内电流表达式为 :
J
nFzc
exp
G0
nF ( RT
平)
c0O
J0
exp
nF( RT
平)
J
nFza
二.电极电位对电子转移步骤绝对反应速
度的影响
O+ne⇋R
根据绝对反应速度理论,上述反应在φ=0
电化学理论与方法 第七章 电子转移动力学
(3)塔菲尔斜率即塔菲尔公式种的b值,由b值可求出 电极反应的传递系数α或β。 (4)在平衡电极电位附近测量极化曲线,该极化曲线 靠近平衡电位的线性部分的斜率即为极化电阻。 (5)由求得的j0值可计算电极反应速度常数。
§6.4多电子的电极反应
1、反应历程的可能性 有两种可能: (1)一次转移两个电子,这种情况发生在高过电位下。 (2)在较低过电位下,一次只转移一个电子,连续进行两 次单电子的电极反应。 这种反应历程称为多电子转移步骤或多步骤电化学反应。 该历程中,依靠连续进行的若干单电子转移步骤完成电化学 反应过程。 2、多电子转移步骤的动力学规律 电流密度与过电位的关系:
0
(6.14)
F j j exp RT
0
(6.15)
对式(6.14)和式(6.15)取对数,经整理得到
2.3RT 2.3RT 0 log j log j F F
(6.16)
2.3RT 2.3RT 0 log j log j F F
(6.17)
§6.3.2 净电流方程(Butler-Volmer方程)
电极过程达到稳态时,电极反应的速度就等于电子转移 步骤的净反应速度(即净电流密度)。
j j净
对单电子电极反应
F F j j净=j exp exp RT RT
0
F j c j exp c RT
0
(6.39)
实质是还原反应速度远大于氧化反应速度。式(6.39) 取对数可得到阴极极化和阳极极化的过电位与电流密度 的关系: 2.3RT 2.3RT
c
F
log j
0
F
log j c
第五章电化学步骤动力学
exp(
G ) RT
氧化反 应速度
j
Fk cR
exp(
G ) RT
(5.12) (5.13)
1、电子转移步骤基本动力学公式推导(2)
• 由于液相传质步骤处于准平衡态,有:
表面 浓度
• 则;
cO cO 溶液体 浓度
cR cR
j
FkcO
exp(
G RT
)
(5.14)
j
FkcR
exp(
氧化态
还原态 4—双电层中电子位能
变化
2—双电层电位差为
时电子的位能曲线
一、电极电位对电子转移步骤活化能的影响(7)
• 由图可见:
G G0 F (5.2)
还原反应活化能增加
G G0 F
(5.3)
氧化反应活化能减少
一、电极电位对电子转移步骤活化能的影响(8)
小结:
有界面电场时,反应粒子达到活化态需要多作 功 nF 。还原反应 氧化反应
FKcO
exp(
F平
RT
)
FKcR
exp(
F平
RT
)
对于单电子反应 1
令 0 RT ln K
整理后得:
FK
平
0
RT F
ln
cO cR
(5.30)
0 0 RT ln O F R
(5.31)
二、交换电流密度与电极反应的动力学特性
3、电极处于非平衡状态时,可用
j0 表示
j、j
j
FKcO
exp
F
RT
(平
)
j0 exp( F )
RT
(5.32)
求解
j
第六章电子转移步骤动力学
一、交换电流密度
1、定义: • 电极反应 当 平
O e R j j 时
log j
即
F平 j FKcO exp( ) RT (6.26) F平 j FKcR exp( ) RT (6.27)
第六章 电子转移步骤动力学
• 电子转移步骤(电化学反应步骤): 反应物质在电极/溶液界面得到电子或失 去电子,从而还原或氧化成新物质的过 程。
• 当电子转移步骤成为电极过程的控制步 骤时,产生电化学极化,整个电极的极 化规律取决于电子转移步骤的动力学规 律。
§6.1 电极电位对电子转移步骤反应速度的影响 §6.2 电子转移步骤的基本动力学参数 §6.3 稳态电化学极化规律 §6.4 多电子的电极反应 §6.5 双电层结构对电化学反应速度的影响 §6.6 电化学极化与浓差极化共存时的动力学规律 §6.7 电子转移步骤量子力学简介
3、举例
• 250C 银浸入
0.74V , j1 10m A/ cm2 F 即 : j1 FKc Ag exp( ) 10m A/ cm2 RT 若电极电位正移 0.24V,则银的溶解速度为: F( 0.24) j2 FKc Ag exp RT F F 0.24 FKc Ag exp exp RT RT F 0.24 2 j1 exp 1000 m A / cm RT
• 将电极电位与活化能的关系代入:
0 G K k exp( ) RT
0,电位坐标零
点处的反应速度常数
(6.16) 0 G F j Fk cR exp( ) RT F FKcR exp( ) RT
化学反应中的电子转移反应动力学研究
化学反应中的电子转移反应动力学研究化学反应中的电子转移反应动力学研究在现代化学领域中扮演着重要的角色。
了解反应速率和能量变化的动力学过程对于设计新的催化剂、预测催化反应的效率以及优化反应条件具有重要作用。
本文将介绍电子转移反应的动力学研究方法与应用。
一、电子转移反应的基本概念电子转移反应是指在化学反应中,电子从一个物质转移到另一个物质的过程。
这种反应通常发生在氧化还原反应中,其中一个物质失去电子(被氧化),而另一个物质获得电子(被还原)。
电子转移反应的动力学研究主要关注反应速率以及反应路径中的能量变化。
二、电子转移反应动力学研究方法1. 光谱学方法光谱学方法是电子转移反应动力学研究中常用的方法。
通过测量反应物和产物之间的吸收、发射或散射光谱,可以获得反应过程中电子能级的信息。
例如,紫外可见吸收光谱、荧光光谱和拉曼光谱等可以用于研究电子转移反应过程中的能量变化以及反应物和产物之间的电子转移路径。
2. 电化学方法电化学方法是另一种用于电子转移反应动力学研究的常用方法。
电化学实验可以通过测量电流和电势的变化来研究反应物和产物之间的电子转移过程。
其中,循环伏安法、计时电流法和旋转圆盘电极法等常用技术可以用于测定电子转移反应的速率常数和电子转移动力学参数。
3. 分子动力学模拟方法分子动力学模拟方法是近年来发展起来的一种重要的电子转移反应动力学研究方法。
这种方法通过在计算机上模拟器件分子系统的动力学过程,可以获得电子转移过程中的能量变化和反应速率常数等信息。
分子动力学模拟方法可以提供原子级别的细节,对于研究复杂的电子转移反应机理非常有价值。
三、电子转移反应动力学研究的应用1. 催化剂的设计与优化电子转移反应动力学研究对于催化剂的设计和优化至关重要。
了解反应的动力学特性可以帮助科学家确定催化剂的合适活性位点和优化催化反应的条件,从而提高反应的选择性和效率。
2. 化学反应速率预测电子转移反应动力学研究可以提供反应速率常数和反应路径的信息,使得科学家能够预测和解释化学反应的速率。
电子转移步骤动力学
三、金属和溶液中电子能级的分布
T 0K 时,EF 附近的能级是部分充满的,只有在这个区 域的电子可以自由移动、参加电极反应。
•通常将 EF 看 作是反应电子的 平均能级;也是 金属中自由电子 的能级;也是自 由电子在金属中 的电化学位。
T 0K T 0K
EF
3、 溶 液 中 的 电 子 能 级 分 布
平衡电位时:
还原反应电子跃迁速度
五、平衡电位下和电极极化时的电子跃迁
• 平衡电位时:
j
j
j0
•电极极化时
EF发生变化
EF0
(O
/
R
不变
)
j j
jc
j
j
ja j j
五、平衡电位下和电极极化时的电子跃迁
• 电极电位影响反应活化能和反应速度的实质 电极极化 EF 移动 电子发生隧道跃迁的条件变化 电子达到激发态所需的能量变化 电子转移步骤的活化能变化 电极反应速度变化
• 前述中,假设电极电位的改变只引起紧密层电
位差发生变化。认为分散层中电位差的变化为
零。
( 1)
• 在稀溶液中、零电荷电位附近和有表面活性剂 吸附时, 1 电位及其变化对电化学反应速度影 响很大,称为 1 效应。
§6.5 双电层结构对电化学反应速度的影响
一、双电层结构对电化学反应速度影响的机理 二、考虑了 1 效应的基本动力学公式 三、 1电位对不同电化学反应的影响
O粒子基态 电子能级
R粒子基态 电子能级
充满
费米能级 F(E)=0.5
价电子的能 级分布函数
3、溶液中的电子能级分布
•
溶液中标准O/R体系的费米能级定义为
E0 F(O/ R)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
三种前提假设: (1) 溶液中参与反应的Ag+离子位于外亥姆荷茨平面 ,电极上参与反应的Ag+离子位于电极表面的晶格中,活 化态位于这二者之间的某个位置。 (2) 电极/溶液界面上不存在任何特性吸附,也不存在 除了离子双电层以外的其他相间电位。也就是说,我们只 考虑离子双电层及其电位差的影响。 (3)溶液总浓度足够大,以致于双电层几乎完全是紧 密层结构,即可认为双电层电位差完全分布在紧密层中, ψ1=0
第六章 电子转移步骤动力学
电子转移步骤(电化学反应步骤)系指反应物质在电极/溶液界 面得到电子或失去电子,从而还原或氧化成新物质的过程。 这一单元步骤包含了化学反应和电荷传递两个内容,是整个 电极过程的核心步骤。因此,研究电子转移步骤的动力学规 律有重要的意义。尤其当该步骤成为电极过程的控制步骤, 产生所谓电化学极化时,整个电极过程的极化规律就取决于 电子转移步骤的动力学规律。对该步骤的深入了解,有助于 人们控制这一类电极过程的反应速度和反应进行的方向。
对式取对数,整理,得电子转移步骤的基本动 力学公式:
对图6.3的解释
Βιβλιοθήκη 曲线1:纯化学反应活化能变化 曲线3:界面电场电压降 曲线4:有界面电场后的电功(nFE) 曲线2:曲线1与曲线4的加和(电化学位)。
另外:对曲线4,分成了α、β两部分,对于还 原反应,为αnFE ,对于氧化反应,为βnFE 。 α+β=1,传递系数。
问题: 有界面电场后,还原反应与氧化反应的活化能(自 由能)有什么变化?
电化学动力学的第一组方程 根据化学动力学,反应速度与反应活化能之 间的关系为:
阿累尼乌斯方程
斯万特·奥古斯特·阿累尼乌斯(Svante August Arrhenius) 瑞典物理化学家,1859年2月19日生于瑞乌普萨拉附近的维克城 堡。电离理论创立者。学术成果:解释溶液中的元素是如何被 电解分离的现象,研究过温度对化学反应速度的影响,得出著 名的阿累尼乌斯公式。还提出了等氢离子现象理论、分子活化 理论和盐的水解理论。对宇宙化学、天体物理学等也有研究。 获得1903年诺贝尔化学奖。
还原反应: 原来: 现在: 氧化反应: 原来: 现在:
=
+ αnFE
= == =
+ αnFE-nFE + αnFE-n(α+β)FE -nβFE
与零电荷电位相比,由于界面电场的影响, 氧化反应活化能减小了,而还原反应活化能 增大了。
小结:电化学动力学的三组基本公式
活化能与电极电位的关系式
1901年,开始首届评选诺贝尔奖的时候,阿累 尼乌斯是物理奖的11个候选人之一,可惜落选 了。1902年他又被提名诺贝尔化学奖,他也没 有被选上。1903年,评奖委员会很多人都推举 阿累尼乌斯,但是,对于他应获得物理奖还是 化学奖发生分歧。诺贝尔化学奖委员会提出给 他一半物理奖,一半化学奖,这一方案过于奇 特,被否定了。又提出他获奖问题延期至第二 年,也被否决。电离学说在物理学和化学两个 学科都具有很重要的作用,人们一时很难确定 他应该获得哪一个奖项。最后,阿累尼乌斯获 得了1903年诺贝尔化学奖。他是第一个获得这 种崇高荣誉的诺贝尔的同胞。
6.1 电极电位对电子转移步骤反应速度的影响
电极过程最重要的特征就是电极电位对电极反应速度的影响。 这种影响可以是直接的,也可以是间接的。当电子转移步骤 是非控制步骤时,如前所述,电化学反应本身的平衡状态基 本未遭破坏,电极电位所发生的变化通过改变某些参与控制 步骤的粒子的表面浓度而间接地影响电极反应速度。例如上 一章中.扩散步骤控制电极过程(浓差极化)时的情形就是如 此。这种倩况下,仍可以借用热力学的能斯持方程来计算反 应粒子的表面浓度。所以,电极电位间接影响电极反应速度 的方式也称为“热力学方式”。当电子转移步骤是控制步骤时, 电极电位的变化将直接影响电子转移步骤和整个电极反应过 程的速度,这种情形称为电极电位按“动力学方式”影响电极 反应速度。本章中所涉及的正是这后一种情形。
电化学动力学的第二组方程
设电极反应为:
注:非单纯的化学反应,而是电化学反应
用电流密度表示的还原反应和氧化反应的速 度,即(j=nFv):
在研究电子转移步骤动力学时,该步骤通 常是作为电极过程的控制步骤的,所以可 认为液相传质步骤处子准平衡态,电权表 面附近的液层与溶液主体之间不存在反应 粒子的浓度差。再加亡已经假设双电层中 不存在分散层。
电化学动力学的第三组方程 注:本方程与活化能相关
该反应为化学反应
化学能变化(化学位)
在电化学体系中,荷电粒子的能量可用电 化学位表示。在零电荷电位时,Ag+离子的 电化学位等于化学位。当电极/溶液界面存 在界面电场时,例如电极的绝对电位为△φ ,且△φ>0时,电化学位用下式表示:
问题: 当存在界面电场时,化学反应变为电化学反应,此时, 界面电场对自由能产生什么样的影响?
图6. 2为银电极刚刚浸入硝酸银溶液的瞬间,或者是零电荷电位 下的Ag+离子的位能曲线。图中O为氧化态(表示溶液中的Ag-离 子),R为还原态(表示在金属晶格中的Ag+离子)。曲线OO’表示 Ag+离子脱去水化膜,自溶液中逸出时的位能变化.曲线RR’表 示Ag+离子从晶格中逸出时的位能变化。二者综合起来就成了 Ag+离子在相间转移的位能曲线OAR。交点A即表示
从化学动力学可知,反应粒子必须吸收一定的能量,激发到 一种不稳定的过渡状态——活化态,才有可能发生向反应产 物方向的转化。也就是说,任何一个化学反应必须具备一定 的活化能.反应才得以实现。上述规律,可以用图6.1形象 地表示出来。 图6.1中ΔGl为反应始态 (反应物)和活化态之间的 体系自由能之差,即正向 反应活化能。ΔG2为反应 终态(产物)与活化态之间 的体系自由能之差,表示 逆向反应活化能。因为一 般倩况下,ΔGl≠ΔG2,故 正向反应速度和逆向反应 速度不等,整个体系的净 反应速度即为二者的代数 和。