液相色谱分析--仪器分析
色谱图仪器分析实验报告

色谱图仪器分析实验报告一、实验目的本实验旨在通过使用色谱图仪器对样品的成分进行分析,掌握色谱仪的操作步骤和原理,提高实验操作技巧,理解色谱技术在分析中的应用。
二、实验原理色谱图仪是一种分离和分析化学物质的仪器。
它的原理是基于物质在色谱柱中通过相互作用后在不同的时间点相继排出,形成色谱图。
常见的色谱技术包括气相色谱(GC)和液相色谱(LC)。
本实验主要以液相色谱为例进行分析。
液相色谱将待测样品溶解于流动相,通过液相流动将样品载送到柱子中,样品与固定在柱子上的固定相发生相互作用,不同成分在相互作用力的影响下以不同的速率通过柱子,并在检测器中形成峰。
检测器可以通过对各个峰进行测量和分析,最终得到样品的成分信息。
三、实验步骤1. 实验前准备准备待测样品及相关试剂,检查色谱仪的操作状态,并进行必要的预热和标定。
2. 溶解样品将待测样品溶解于溶剂中,并进行适当的稀释,使样品的浓度满足分析要求。
3. 注射样品使用微量注射器,将稀释后的样品注射到色谱柱中。
4. 色谱条件设置设置色谱柱温度、流动相流速和组成,以及检测器参数。
5. 开始分析点击色谱仪上的开始按钮,开始流动相的流动,观察样品在色谱柱中的分离情况,记录检测器上出现的峰的数目和峰的形状。
6. 数据处理使用数据处理软件对收集到的数据进行峰面积、峰高等参数的测量和计算,绘制色谱图。
四、实验结果与讨论在本次实验中,我们以某种药物作为待测样品,通过色谱仪进行分析。
根据实验步骤,我们成功地将溶解后的药物样品注射到色谱柱中,在设定的色谱条件下进行了分析。
在观察色谱图的过程中,我们发现在某个特定的时间点,药物样品在检测器上形成了一个明显的峰。
根据峰的形状和峰的位置,我们可以初步判断药物样品中的化学成分。
通过数据处理软件进行峰面积、峰高等参数的测量和计算,我们得到了更精确的分析结果。
根据峰面积的大小,我们可以推测药物样品中不同成分的含量。
然而,在实验过程中我们也遇到了一些困难。
仪器分析 高效液相色谱法

第17章HPLC法17.1 内容提要17.1.1 基本概念高效液相色谱法──在经典液相色谱法的基础上,引入了气相色谱(GC)的理论,在技术上采用了高压泵、高效固定相和高灵敏度检测器,使之发展成为高分离速率、高分离效率、高检测灵敏度的高效液相色谱法,易称为现代液相色谱法。
高效液相色谱仪──采用了高压输液泵、高效固定相和高灵敏度检测器等装置的液相色谱仪称为高效液相色谱仪。
梯度洗脱──用两种(或多种)不同极性的溶剂,在分离过程中按一定程序连续的改变流动相的浓度、配比和极性,使样品中各组分能在最佳的分配比下出峰的操作技术。
也称为梯度淋洗。
低压梯度──又称外梯度,特点是先混合后加压。
它是采用在常压下预先按一定的程序将溶剂混合后再用泵输入色谱柱系统,易称为泵前混合。
高压梯度──又称内梯度,特点是先加压后混合。
它有两台高压输液泵、梯度程序器(或计算机及接口板控制)、混合器等部件组成。
两台泵分别将两种极性不同的溶剂输入混合器,经充分混合后进入色谱柱系统,是一种泵后高压混合形式。
柱外效应──由色谱柱以外的因素引起的色谱峰形扩展的效应。
柱外因素常指从进样口到检测器之间,除色谱柱以外的所有死时间,如进样器、连接管、检测器等的死体积,都会导致色谱峰形加宽、柱效下降。
液固吸附色谱法──以固体吸附剂为固定相,吸附剂表面的活性中心具有吸附能力,样品分子被流动相带入柱内,它将与流动相溶剂分子在吸附剂表面发生竞争吸附性。
K值大的强极性组分易被吸附,K值小的弱极性组分难被吸附,样品组分因此被分离。
液液分配色谱法──根据物质在两种互不相溶(或部分互溶)的液体中溶解度的不同,有不同的分配,从而实现分离的方法。
分配系数较大的组分保留值也较大。
正相分配色谱法──流动相极性低而固定相极性高的称为正相分配色谱法。
反相分配色谱法──流动相极性高而固定相极性低的称为反相分配色谱法。
化学键合相──利用化学反应将有机分子键合到载体表面上,形成均一、牢固的单分子薄层而形成的各种性能的固定相。
《仪器分析》第八章 液相色谱分析

Si Cl
+ R H N 2
Si N R H
热和化学稳定性比酯型好。 热和化学稳定性比酯型好。
Si C l
+ R M X g
Si R
从理论上讲,这种结构具有更好的稳定性, 从理论上讲 , 这种结构具有更好的稳定性 , 特别是对于微碱性流动相, 而且R基可以多 特别是对于微碱性流动相 , 而且 基可以多 次氯化,形成聚烷基键合相, 但是制备困难。 次氯化 , 形成聚烷基键合相 , 但是制备困难 。
柱体为直型不锈钢管,内径1~ 柱体为直型不锈钢管,内径 ~6 mm, , 柱长5~ 柱长 ~40 cm。发展趋势是减小填料 。发展趋势是减小填料 粒度和柱径以提高柱效。 粒度和柱径以提高柱效。 以提高柱效
(7)检测器 检测器
♥ 紫外检测器
70%应用,氘灯,适用于梯度淋洗 对流动相速度变化不敏感 溶剂选择时紫外检测器波长不能小于溶剂的紫外截至波 长;
(5)进样器 进样器
♥ 高压进样阀-用微量注射器将样品注入样品环管(10µL到 2mL)
(6)色谱柱 色谱柱
♥ 色谱系统的心脏 ♥ 优质不锈钢管,内壁光洁平滑 ♥ 接头死体积尽可能小 ♥ 为了保护色谱柱不被污染,有时候需要在分析柱前加一根 短柱,称为卫柱。为了防止由于卫柱而过分增加压力,在 卫柱中使用的颗粒大小约10-30 µm。
3. Si-O-Si-C键型(硅胶和有机硅烷的反应)
R Si O + R SiX H 3 Si O Si R R
目前用得最多的类型。 目前用得最多的类型 。 具有良好的热合 化学稳定性, 能够在pH2~8.5的介质中 化学稳定性 , 能够在 的介质中 使用。 使用。
仪器分析-高效液相色谱法

流动相的选择与制备
选择合适的流动相
根据被分析化合物的性质, 选择适当的流动相,如有 机溶剂、缓冲液等。
流动相的配制
按照实验要求,准确称量 流动相组分,混合均匀, 并进行过滤和脱气处理。
流动相的梯度洗脱
对于多组分分离,可以采 用梯度洗脱技术,以提高 分离效果。
仪器的开机与平衡
开机
按照仪器说明书,打开仪器电源, 启动仪器操作系统。
药物制剂质量控制
高效液相色谱法可以用于药物制剂的质量控制, 检测制剂中药物的含量、纯度和稳定性等指标。
环境样品分析中的应用
污染物检测
高效液相色谱法可以用 于检测环境中的有机污 染物,如农药、多环芳 烃等,为环境污染控制 和治理提供依据。
饮用水质量检测
通过高效液相色谱法可 以检测饮用水中的有害 物质,如消毒副产物、 微量有机物等,保障公 众的饮用水安全。
粒径
色谱柱的粒径影响分离效 果和分离时间。粒径越小, 分离效果越好,但分离时 间越长。
长度
色谱柱的长度影响分离效 果和载样量。长度越长, 分离效果越好,但载样量 越小。
检测器
类型
常用的检测器有紫外-可见光检测器、荧 光检测器、电导检测器等,根据被测物质 的性质和检测需求选择合适的检测器。
响应速度
线性范围
质。
测定水体、土壤、空气 中的污染物和有害物质。
用于蛋白质、核酸、细 胞等生物大分子的分离
和检测。
高效液相色谱法的优势与局限性
优势
高分离效能、高灵敏度、高选择 性、应用范围广。
局限性
需要专业操作人员、仪器昂贵、 样品前处理复杂、耗时长。
02 高效液相色谱法的仪器构成
CHAPTER
仪器分析9-经典液相色谱法概要

2.液相色谱的固定相和流淌相
3〕常用有机吸附剂
① 聚酰胺 为高分子聚合物质,不溶于水、甲醇、乙
醇等常用有机溶剂,对碱较稳定,对酸稳定 性较差,可溶于浓盐酸、冰醋酸及甲酸。
2.液相色谱的固定相和流淌相
聚酰胺对有机物质的吸附属于氢键吸附,通 过分子中的酰胺羰基与酚类,或酰胺键上的游 离氨基与醌类、脂肪羧酸上的羰基形成氢键缔 合而产生吸附。吸附的强弱则取决与各种化合 物与之形成氢键缔合的力量。
2.液相色谱的固定相和流淌相
2〕常用无机吸附剂
① 硅胶〔SiO2·H2O〕
硅胶为极性吸附剂,外表主要存在着硅羟基〔硅 醇基〕和暴露于外表的Si-O-Si键,另外还有一些硅 醇基可能与水以氢键键合。硅羟基的外表浓度在吸 附色谱中很重要,由于人们通常认为硅羟基是强吸 附位点,而Si-O-Si则是疏水性的。
氧化铝的活性与其含水量相关。
2.液相色谱的固定相和流淌相
氧化铝适宜分别溶于有机溶剂的极性、弱极 性的非强离解型的化合物,尤其适合于分别芳 香族化合物。当样品为碱性化合物时,用硅胶 分别会造成严峻吸附,此时可选用氧化铝进展 分别,但酸性易离解的化合物简洁在氧化铝上 形成死吸附。
氧化铝分别几何异构体力量优于硅胶。
2.液相色谱的固定相和流淌相
(3) 离子交换树脂的性质 1) 离子交换树脂的特性
二乙烯苯
重量交联度:树脂中所合交联剂的百分率。
树脂的交联度越大,则网眼越小,交换时体积大 的离子进入树脂便受到限制。但提高了交换的选择 性;另外,交联度大时,形成的树脂构造严密,机 械强度高。但是假设交联度过大则对水的膨胀性能 差,交换反响的速度慢,因此要求树脂的交联度一 般为8-12%。
2.液相色谱的固定相和流淌相
仪器分析-色谱法

高效液相色谱法(HPLC) 是在气相色谱和经典液相色谱的基础上,采用高压泵、高效固定相以及高灵敏度检测器等新实验技术建立的一种液相色谱分析法。
特点:高压、高柱效、高灵敏度2.HPLC中分离条件的选择:a.固定相与装柱方法的选择:选粒径小的、分布均匀的球形固定相(dp≤10μm)首选化学键合相,匀浆法装柱b.流动相及其流速的选择: 选粘度小、低流速的流动相c.柱温的选择:选室温25-30℃左右。
太低流动相黏度增加,太高容易产生气泡第一节液-固色谱法1.液-固色谱法是利用各组分在固定相上的吸附能力不同进行分离的,也称液-固吸附色谱。
2.分离原理.:组分分子与流动相分子竞争吸附吸附剂表面活性中心,靠组分分子的分配比不同而分离。
3.吸附剂吸附试样的能力,主要取决于吸附剂的比表面积和理化性质,试样的组成和结构以及流动相的性质等。
1)组分与吸附剂的性质相似时,易被吸附;2)组分分子结构与吸附剂表面活性中心的刚性几何结构相适应时,易于吸附。
吸附色谱是分离几何异构体的有效手段;不同的官能团具有不同的吸附能力,因此,吸附色谱可按族分离化合物4.固定相:常用的液-固色谱固定相是表面多孔和全多孔微粒型硅胶、氧化铝等。
一般采用5~10μm的全多孔型微粒。
这些吸附剂的极性都比较大,对非极性组分的保留能力较弱,与极性化合物的相互作用较强。
5.流动相:在液-固色谱中,选择流动相的基本原则是极性大的试样用极性较强的流动相,极性小的则用低极性流动相。
液-固色谱的流动相必须符合下列要求:1)能溶解样品,但不能与样品发生反应。
2)与固定相不互溶,也不发生不可逆反应。
3)粘度要尽可能小,这样才能有较高的渗透性和柱效。
4)应与所用检测器相匹配。
例如利用紫外检测器时,溶剂要不吸收紫外光。
5)容易精制、纯化、毒性小,不易着火、价格尽量便宜。
第二节化学键合相色谱法1.液液分配色谱法分离原理:根据物质在两种互不相溶的液体中溶解度的不同,在两溶液间进行不同分配而实现分离。
仪器分析第4讲 高效液相色谱法

经典液相色谱法 75-600 0.01-1.0 1-20 50-200 2-50 1-10
高效液相色谱法 3-50(常用5-10)
20-300 0.05-1.0
2-30 104-105 10-6-10-2
2.高效液相色谱法与气相色谱法
(l)气相色谱法分析对象只限于分析气体和 沸点较低的化合物,它们仅占有机物总数 的20%.对于占有机物总数近80%的那些高 沸点、热稳定性差、摩尔质量大的物质, 目前主要采用高效液相色谱法进行分离和 分析.
3. 柱外效应
由于色谱柱之外的因 素引起的色谱峰的展 宽,例如进样系统、 连接管路及检测器的 死体积等。
3-3 高效液相色谱的类型及其分离原理
液—液分配色谱及化学键合相色谱 液—固吸附色谱 离子交换色谱 离子色谱 空间排阻色谱
1、 液-液分配色谱
liquid- liquid partition chromatography
4、 离子色谱
ion chromatography
离子色谱法是由离子交换色谱法派生出来的一种 分离方法。由于离子交换色谱法在无机离子的分 析和应用受到限制。例如,对于那些不能采用紫 外检测器的被测离子,如采用电导检测器,由于 被测离子的电导信号被强电解质流动相的高背景 电导信号掩没而无法检测。
2、 液-固吸附色谱
liquid-solid adsorption chromatography
流动相为液体,固定相为固体吸附剂
分离原理:利用溶质分子占据固定相表面吸附 活性中心能力的差异
分离前提:K不等或k不等
液—固吸附色谱
固体吸附剂主要类型: 极性的硅胶(应用最广) 氧化铝 分子筛 非极性的活性炭
1971年科克兰等人出版了《液相色谱的现代实践》一 书,标志着高效液相色谱法(HPLC)正式建立。
仪器分析笔记 《高效液相色谱分析》
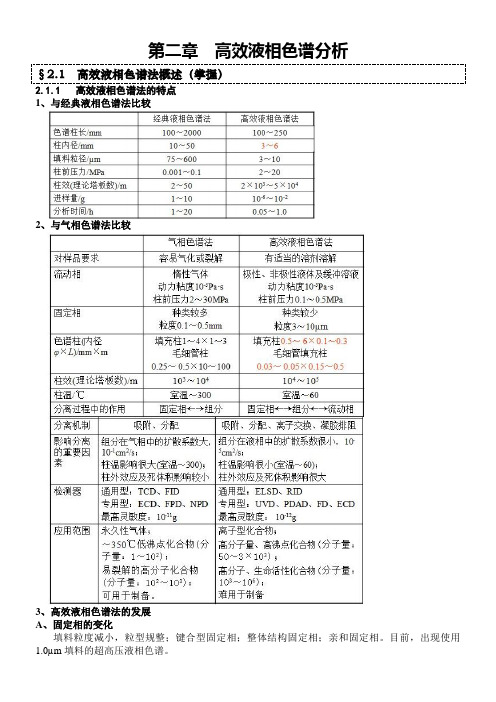
第二章高效液相色谱分析§2.1 高效液相色谱法概述(掌握)2.1.1 高效液相色谱法的特点1、与经典液相色谱法比较2、与气相色谱法比较3、高效液相色谱法的发展A、固定相的变化填料粒度减小,粒型规整;键合型固定相;整体结构固定相;亲和固定相。
目前,出现使用1.0µm填料的超高压液相色谱。
B 、流动相变化目前,出现120~220℃超热水为流动相、FID 和FPD 检测器的HPLC 。
C 、全新方法剪切流路液相色谱;不同分离机制组合的多维液相色谱以及HPLC 与MS 、NMR 、IR 联用的多维液相色谱法。
4、高效液相色谱法的特点①高压 :采用高压输液设备,(150~350)× 105 Pa ②高速:分析速度快; ③高效:柱效很高。
(n>30000),可以在数分钟内完成数百种物质的分离;④高灵敏度:10—9g (UV );10—11g (荧光检测)。
5、高效液相色谱法的局限①溶剂用量太大;②缺乏诸如气相色谱使用的TCD 、FID 通用型检测器; ③不能替代气相色谱法,难分离化合物(柱效10万以上),必须使用毛细管气相色谱法进行分离; ④不能替代中、低压柱色谱法,一些生物活性化合物不能承受200kPa ~1MPa 压力。
§2.2 影响色谱峰扩展及色谱分离的因素(了解)2.2.1 影响色谱峰的扩展的因素高效液相色谱法的基本概念及理论基础,与气相色谱法是基本一致的,其区别主要在于流动相的不同。
现根据速率理论及色谱峰扩展及色谱分离的影响讨论如下:高效液相色谱的范氏方程:2222m p sm p s fd m p mm s C d C d C d C D H d u u u D D D λ⎛⎫=++++ ⎪ ⎪⎝⎭ 若将上式简化,可写作:BH A Cu u=++这与气相色谱的速率方程形式是基本一致的,主要区别在于纵向扩散项可以忽略不计,影响柱效的主要因素是传质项。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
液相色谱分析法
-------复方阿司匹林有效成分的分析
姓名:高伟
班级:环工0801
学号:200829090119
知识准备:
复方阿斯匹林由阿斯匹林、非那西汀和咖啡因三种药物组成。
阿司匹林分子式为C9H8O4,非那西汀,分子式为:C10H13NO2。
咖啡因C8H10N4O2。
容量分析法、胶束薄层色谱法、PLS-紫外分光光度法、区带毛细管电泳法分别对三个成分进行了成功测试,研究发现液相色谱分析法可以很好的分析复方阿司匹林的各种成分。
实验过程:
a) 实验原理
由于阿司匹林类药品在生产和储运过程中容易吸潮分解生成水杨酸,此外样品在溶解和分析过程中有时也有降解现象,因此在APC片的分析测试中不可忽略其水解产物水杨酸的干扰因素。
HPLC 技术在药物分析中有很多应用,以往对APC 的测定很少考虑其降解因素。
本文实验发现,用二氯甲烷-乙腈混合溶剂溶解样品,在甲醇-水体系中加入少量乙酸和磷酸作为流动相,既能有效避免阿司匹林的进一步降解,又可以将三个组分与水杨酸很好分离,据此建立的HPLC 法可以同时测定APC 片中各种成分。
b) 仪器和试剂
岛津LC-10A高效液相色谱仪;十八烷基键合固定相色谱柱(岛津VP-ODS 150Lx4.6);20μL定量进样管;紫外检测器;甲醇和乙腈为色谱纯;二氯甲烷、乙酸和磷酸为分析纯;APC和水杨酸的对照品为分析纯;复方阿司匹林片。
c) 实验步骤
1) 混合对照品标准溶液的制备
按处方配比准确称取阿司匹林0.2268g、咖啡因0.0350g和非那西汀0.1620g,置于同一干净烧杯中,用二氯甲烷-乙腈(V:V=3:2)溶解后转入500mL容量瓶中,稀至刻度制成浓度为0.8476g ·L-1的混合对照品标准溶液。
其中阿司匹林、咖啡因和非那西汀的浓度分别为0.4536g ·L-
1、0.0700g ·L-1和0.3240g ·L-1。
2) 样品储备溶液的制备
将准确称重的市售APC片20片(9.8741g)于乳钵中研细混匀,准确称取相当于1片的重量(0.4937g)置于干净烧杯中,用适量二氯甲烷-乙腈(V:V=3:2)充分溶解并滤除残渣后,转移至500mL容量瓶中稀至刻度。
色谱条件的选择流动相是甲醇—水—乙酸—磷酸,体积比
46:52:1.5:0.5;紫外检测波长为279nm;柱温35℃;洗脱速度为0.8 mL·min -1。
3) 测定方法
用注射器将适量待测物溶液(多于20μL)注入定量管,通过六通阀切入色谱流路进行分离测定,以色谱峰面积进行外标法定量。
d) 结果和讨论..
线性关系和精密度
将0.8476 g ·L-1的混合对照品标准溶液分别稀释成150.0、80.0、50.0、25.0、10.0、5.0、2.5、1.0、.. 0.5μg·mL-1的标准系列溶液,在选定的色谱条件下进
样测定;由于混合对照品溶液中阿司匹林、非那西汀和咖啡因的质量浓度比为0.2268:0.162:0.035,据此可以准确算出标准系列中三个组分的准确质量浓度,分
别以各组分的峰面积对其质量浓度做回归方程可以发现都有很好的线性关系。
此外,用10μg ·mL-1的标准系列溶液连续平行测定12次,计算其相对标准偏差。
各组分的线性关系和精密度
准确度
用混合对照品标准液配制3个不同浓度的标准样品,用以上建立的方法进行测定
从表中可见,阿司匹林的回收率为98.56%~101.57%,平均值为
99.84%;非那西汀的回收率为98.90%~101.57%,平均值为100.29%;咖啡因的回收率为99.23%~101.51%,平均值为100.57%。
表明方法的准确度很好。
e) 实际样品测定
准确移取4.00mL样品储备溶液于250mL容量瓶中稀释至刻度制成样品测试溶液进行分析测定,用外生命科学仪器2005 第3卷/第6期标法定量。
平行测定五次的平均结果见表3所示。
样品中组分含量的计算公式为:
市售正痛片的测定结果
由表可见,咖啡因和非那西汀的测定含量与处方含量十分接近,而阿司匹林的测定含量明显低于处方含量。
这主要是由于阿司匹林部分降解后转变成了水杨酸,致使阿司匹林的含量减少。
在样品的色谱图中可以明显看到水杨酸的存在。
此外,同样用外标法对水杨酸进行定量分析,测得水杨酸含量为15.1 mg·片 -1。
实验讨论
1样品溶剂
通过对APC片在多种溶剂中溶解性和稳定性的比较发现,二氯甲烷-乙腈(V:V=3:2)溶液对APC片的溶解性和稳定性均较好,实验表明,在48h内测定样品溶液,各组分峰的积分面积数值基本不变,可以认为阿司匹林无明显的降解现象。
2流动相
反相色谱中常用的流动相是甲醇-水体系和乙腈-水体系。
本实验发现,阿司匹林在中性含水体系中水解严重,而在酸性含水体系中水解现象可以得到抑制。
本实验以甲醇—水-乙酸-磷酸(体积比
46.0:52.0:1.5:0.5)为流动相,既可使各组分有很好的分离度,又能保证组分在2小时内无水解现象。
3.检测波长
用三个组分的对照品进行紫外扫描可见,三个组分的最大吸收波长各不相同;而在279nm处三个组分的峰型相对比较适中,虽然此时三个组分皆非最大吸
收,但能较好地兼顾各组分的同时测定。
应用范围
液相色谱法的应用:高效液相色谱分析法更适宜于分离,分析高沸点.热稳定性差,生理活性以及相对分子质量较大的物质,因而应用于核酸,肽类,内酯,稠环芳香烃,高聚物,药物,人体代谢产物,生物大分子,表面活性剂,抗氧剂等的分析,在化工,环保临床药物等领域广泛应用,目前在生命科学中又显示出其突出的地位。