气相色谱法测定萘含量知识点解说.
用气相色谱分析法来测定 煤焦油中萘的含量

气相色谱分析法测定煤焦油中的萘摘要:介绍了一种快速准确测定煤焦油中萘含量的气相色谱分析法(GC法)。
与常用的蒸馏结晶法相比,GC法对煤焦油中的萘含量测定可靠、快速、对色谱分析干扰少,灵敏度高,且操作简单、易掌握,适用于企业生产在线控制。
关键词:煤焦油;萘;气相色谱分析;蒸馏结晶法萘的简介:萘是最基本的稠环芳烃,是重要的有机化工原料,主要用于合成苯酐、萘酚、甲基酚、乙基酚、a-萘酐、H-酸、减水剂、分散剂、2,6-二烷基萘、2,6-萘二甲酸、聚萘二甲酸乙二醇(PEN)等。
萘按生产原料的不同分为煤焦油萘和石油萘,目前无论国内外,煤焦油萘都占大多数。
石油萘通常先从石油裂解C10中采用萃取及吸附的方法提取,最好用溶剂吸收洗涤或升华结晶法提纯。
石油萘含硫量低,更适合于生产精萘、精甲基萘等。
萘的作用:萘的应用领域大都集中在生产合成纤维、橡胶、树脂、染料及炸药、农药等工业部门。
1:前言煤焦油中萘含量的测定是指导煤焦油深加工的一个非常重要的指标,可对工业萘的回收率+生产操作物料平衡的计算提供依据。
山东民生煤化有限公司(简称民生煤化)原料煤焦油中萘含量的分析一直采用蒸馏结晶法,该法不属于国家标准,是焦化企业作为中间控制(指导生产的一种较简单的测定方法,需将原料煤焦油预先蒸馏,切割轻油、酚油、萘油、洗油、蒽油5种馏分,然后将前4个馏分熔化混合在一起$按测定洗油萘含量的分析方法,测结晶点,计算含萘量, 由于分析结果为多次测定之和,误差较大,且繁琐,费时。
建立一种快速而灵敏的分析方法是十分必要的,民生煤化采用GC-2001型气相色谱仪可快速准确测定煤焦油中的萘含量。
2:实验部分仪器:GC-2001型气相色谱仪;N-2000双通道色谱数据工作站;StarNX-350型打印机,气相色谱仪分析法是国家标准分析方法,用十二烷作为溶剂,煤焦油中的萘被溶剂萃取后,对萃取液进行色谱分析,再在同样条件下对萘标样进行色谱分析,根据样品中的萘峰和标样中的萘峰峰高计算萘含量, 试验中需用标样先求出校正因子,然后再在同样条件下对样品进行分析N-2000双通道色谱数据工作站会自动对数据进行处理,直接计算出煤焦油的萘含量。
洗油萘含量的测定方法

洗油萘含量的测定方法萘是石油精制工序中的一种重要的部分,也是洗油过程中的污染物之一,其含量的测定对于洗油过程的控制有着重要的意义。
因此,对萘的含量进行测定是洗油生产过程中的一个重要技术环节。
本文以洗油萘含量的测定方法为研究主题,结合相关文献,就洗油萘含量的测定方法进行阐述。
第一部分:萘含量的测定原理萘是洗油过程中的一种污染物,其含量的测定对于洗油生产过程的控制有着重要的意义。
为了精确测定洗油中萘的含量,可采用气相色谱法,是一种在有机溶剂中分离和测量萘的有效方法。
该方法是以洗油萘含物为样品,在一定的温度条件下,通过采用恒定的温度以及配备有可调整恒定比率的气相色谱仪将萘中的各种组分分离出,并用幅值表示分离出的萘含量,从而测定其含量。
第二部分:萘含量的测定步骤1.样品的配制:首先,将洗油萘样品置于气体瓶中,然后用空气把样品的温度升至一定的温度(控制在40℃-50℃之间);2.样品的注入:将样品置于气相色谱仪的样夹中,并在最佳的进样压力下注入样品,样品以有限的流速进入仪器;3.样品的分离:根据恒定比率,样品经过旋转色谱柱后,会出现萘含量的分离,并用幅值表示;4.数据储存:其次,将萘中分离出的各组分的浓度以及分离出的萘含量的幅值数据,储存在电脑中;5.数据分析:最后,对萘的含量的分离数据进行多元线性回归分析,以确定洗油萘含量。
第三部分:萘含量测定的注意事项1.相色谱仪在使用时应严格控制温度,以确保测量结果准确;2.相色谱仪测量过程中,应注意样品的注入速率,以避免样品沉积;3.相色谱仪测量过程中,应注意进样压力,以避免影响测量精度;4.进行数据处理时,应注意保持实验数据的完整性,并确保测量结果的准确可靠。
综上所述,洗油萘含量的测定方法是采用气相色谱仪,以洗油萘样品为样品,在一定的温度条件下,通过采用恒定的温度以及配备有可调整恒定比率的气相色谱仪将萘中的各种组分分离出,并用幅值表示分离出的萘含量,从而测定其含量。
气相色谱法检测浓氨水中萘含量的试验研究

70柳钢磷酸洗氨工序生产的浓氨水由管道送入烧结机机头进行烟气脱硫。
由于浓氨水中的萘容易结晶从而堵塞管道,须对其含量进行测定,从而为生产工艺调节提供依据。
由于浓氨水中萘只是微量存在,在分析了萘的物理特性之后,在没有液相色谱仪的情况下采用气相色谱法进行试验,验证了气相色谱法也可以进行浓氨水中萘含量的检测。
1 样品预处理使用萃取–分离的方法,利用溶剂的相似相溶原理,使用有机溶剂将萘从浓氨水中分离后再进行色谱测定。
因我厂浓氨水还需要分析含油项目,该项目使用四氯化碳(CCl 4)作为萃取剂,因此,本试验优先试用四氯化碳作为萃取剂进行试验。
在定量的浓氨水中加入一定量的CCl 4,摇匀后静置分层,取下层进行试验。
2 气相色谱试验2.1 试验原理利用固定相中各挥发物的分配系数不同,样品经气化,由载气带入色谱柱,在柱内进行反复分配,混合的各组分实现分离,并先后通过检测器产生信号,形成色谱峰。
将标准样品色谱峰与试样色谱峰进行对比来定量测定[1]。
2.2 试验样品准备2.2.1 纯萘(99.25%分析纯化学试剂)样品称取0.05g纯萘样品放入100mL注射器中,在180℃烘箱中使萘全部升华并保持温度避免其结晶。
2.2.2 纯萘的CCl 4溶液标准样品准确称取0.0500g 纯萘,溶解于C C l 4中,定溶至50mL,再取该溶液5mL,稀释至50mL,此标样的萘含量以1000mL氨水萃取至50mL计,含量为5mg/L。
2.3 样品试验2.3.1 纯萘样品试验进行纯萘样品试验,主要目的是找出在该色谱仪测定条件下萘的出峰时间、峰形状等信息。
纯萘–空气样品在气化后,趁热以1000μL注射器将其进样,观察其出峰情况,在7.5~10min时有一样品峰出现。
2.3.2 纯萘的CCl 4溶液样品试验将5m g /L 四氯化碳标准样品用10μL 的注射器进样,观察其出峰情况,进样初期有一高峰出现,在7.5min~10min时有一样品峰,可判断CCl 4与萘在色谱柱中成功实现分离,进样初期高峰为CCl 4峰,7.5min的样品峰即为萘峰,该色谱柱在既定的试验条件下可以成功实现萘与其它组分分离。
萘含量的测定方法 气相色谱法

萘含量的测定方法气相色谱法警告——使用本标准的人员应有正规实验室工作的实践经验。
本标准并未指出所有可能的安全问题。
使用者有责任采取适当的安全和健康措施,并保证符合国家有关法规规定的条件。
1 范围本标准规定了气相色谱法测定萘含量的试剂、仪器、试样制备、分析步骤及分析结果处理。
本标准适用于精萘和工业萘的定量分析。
2 规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。
凡是注日期的引用文件,其随后所有的修改(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本标准。
GB/T 9722-2006 化学试剂气相色谱法通则3 总则本标准应用了以下原理。
试样被汽化后,随同载气进入色谱柱,各组分经色谱柱得到分离,用火焰离子化检测器(FID)检测,以内标法进行定量分析。
4 试剂和材料除非另有规定,应使用分析纯试剂。
4.1 正己烷:在检测前均应做空白试验以检查试剂的纯度,色谱图上应没有干扰检测的杂质峰。
4.2 无水硫酸铜:在300℃高温炉中灼烧3 h,冷却后保存于干燥器中。
4.3 萘:色谱纯,作标样用。
4.4 正十六烷:色谱纯,作内标用。
5 仪器和设备5.1 气相色谱仪:配有火焰离子化检测器(FID),应符合GB/T 9722-2006中第6章的要求。
5.2 分析天平:精确至0.0001 g。
5.3 恒温水浴:保持温度85℃~90℃。
6 试样制备称取样品30 g~40 g置于洁净干燥的烧杯中,然后将烧杯置于85℃~90℃的恒温水浴中使样品完全融解。
向烧杯中加入无水硫酸铜,直至加入无水硫酸铜不变色为止(约需2 g无水硫酸铜),静置脱水5 min 。
移取溶解的上层样品于内置锡纸的洁净干燥的烧杯中,置于干燥器中冷却。
将冷却后的样品用研钵研成粉末,作为试样检测。
注:对于不含水的样品可直接研成粉末检测,但应在结果中注明。
煤气中萘含量测定方法

煤气中萘含量测定方法(苦味酸法)1 苦味酸法 1.1 原理煤气中萘系物(含萘、甲基萘等) ,在通过苦味酸溶液时生 成结合物沉淀,将过滤后的沉淀溶于丙酮中,用标准碱液滴定, 但煤气中含有茚等某些不饱和烃也能部分地与苦味酸生成结合 物沉淀,以一氯化碘溶液加以校正。
在测定中控制一定温度,并 在测定结果中加上相应校正值,以求得正确的粗萘含量。
1.2 试剂和材料除非另有说明, 在分析中仅有使用确认为分析纯的试剂和蒸 馏水或去离子水或相当纯度的水。
硫酸(H2SQ):密度为 1.84g/mL,含量 95%-98%苦味酸 (2 、4、6 三硝基酚 ) 〔C6H2QH (NQ2) 3〕;e )乙酸铅〔Pb (CHCQQ )・ 3HQ 〕化学纯;f ) 碘化钾( KI ); g) 丙酮( CH3CQC3H ); h ) 冰乙酸( CH3CQQ )H ; i ) 一氯化碘( ICl )化学纯; j ) 可溶性淀粉;k ) 溴百里香酚蓝( C27H28Q5Br 2S ) ;b) 氢氧化钠( NaQH );c) 硫代硫酸钠(N Q S Z Q • 5H 2Q ;a)d)I )硫酸溶液(5f 100):量取5mL 硫酸,缓缓注入约70mL水中,冷却,稀释至 100mL ;m )乙酸铅溶液(50g/L ):称取5g 乙酸铅,溶于70mL 水中,加1mL 冰乙酸,用水稀释至100mL ;n ) 一氯化碘溶液:称取25g 一氯化碘液体,倒入1500mL 冰水中,稀释至 1000mL ;c(NaOH)=0.1moI/L 〕: 按GB/T 601-2002 中 4.1 制备; q ) 硫代硫酸钠标准滴定溶液〔 c (Na2s2o3)=0.05moI/L 〕:按GB/T 601-2002 中 4.6 稀释一倍制备;r )苦味酸溶液:将1瓶25g 的苦味酸溶解在2000mL 蒸馏水中,煮沸,冷却,过滤,将其澄清液用氢氧化钠标准滴定溶液c (NaOH )=0.1moI/L 〕滴定,配制成下列浓度: 1)洗涤液〔c (苦味酸)=0.02mol/L 〕;2) 13C 〜18C 的吸收液〔c (苦味酸)=0.042moI/L 〕; 3) 0 C 的吸收液〔c (苦味酸)=0.033moI/L 〕;吸收过萘的苦味酸溶液可汇集后煮沸、浓缩、冷却、过滤, 将其澄清液再配置成苦味酸溶液〔c (苦味酸)=0.033moI/L 〕或 〔C (苦味酸)=0.042moI/L 〕,重新使用。
固相微萃取-气相色谱法测定煤气中萘

固相微萃取-气相色谱法测定煤气中萘魏华【摘要】The gas was collected with the special sampling bottle. Then, the content of naphthalene in gas was de-termined by gas chromatography( GC) after pretreatment with solid phase microextraction ( SPME) technology. The optimal extraction conditions of SPME were obtained by experiments: the 100 μm polydimethylsiloxane( PDMS) was used as extraction coating;the extraction time at rotate speed of 800 r/min was 5 min at room temperature;the desorption time of gas chromatography was 3 min. The baseline was smooth and steady in presence of acetone sol-vent peak. The determination of naphthalene was not affected by the acetone peak. The results showed that the con-tent of naphthalene exhibited good linearity in range of 5-200 mg/m3 with correlation coefficient of 0. 999 9. The detection limit of method was 0. 08 mg/m3 . The proposed method was applied to determination of naphthalene in gas. The relative standard deviation (RSD, n=7) was 2. 6%-4. 9%, and the recoveries were between 85% and 108%.%采用特制采样瓶收集煤气后,应用固相微萃取( SP ME )对样品进行前处理,采用气相色谱( GC )对煤气中的萘进行测定。
气相色谱法测定萘含量知识点解说
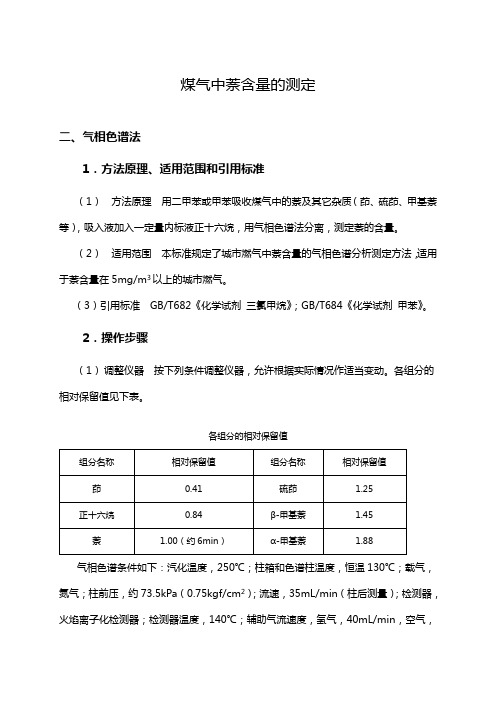
煤气中萘含量的测定二、气相色谱法1.方法原理、适用范围和引用标准(1)方法原理用二甲苯或甲苯吸收煤气中的萘及其它杂质(茚、硫茚、甲基萘等),吸入液加入一定量内标液正十六烷,用气相色谱法分离,测定萘的含量。
(2)适用范围本标准规定了城市燃气中萘含量的气相色谱分析测定方法,适用于萘含量在5mg/m3以上的城市燃气。
(3)引用标准GB/T682《化学试剂三氯甲烷》;GB/T684《化学试剂甲苯》。
2.操作步骤(1)调整仪器按下列条件调整仪器,允许根据实际情况作适当变动。
各组分的相对保留值见下表。
各组分的相对保留值气相色谱条件如下:汽化温度,250℃;柱箱和色谱柱温度,恒温130℃;载气,氮气;柱前压,约73.5kPa(0.75kgf/cm2);流速,35mL/min(柱后测量);检测器,火焰离子化检测器;检测器温度,140℃;辅助气流速度,氢气,40mL/min,空气,400mL/min ;灵敏度和衰减的调节,在萘的绝对进样量为2.5×10-8g 时,产生的峰高不低于10mm ;记录仪纸速,1㎝/min 。
(2)校准①标准样品的制备正十六烷标准溶液:称取7.5g 正十六烷(称准至0.0002g ),置于50mL 容量瓶中,用二甲苯稀释至刻度,混匀,密封贮存备用,溶液浓度应定期检查。
萘标准溶液:称取7.5g 萘(称准至0.0002g ),置于50mL 容量瓶中,用二甲苯稀释至刻度,混匀,密封贮存备用。
校准用标准样品系列的制备:在6个50mL 的小口试剂瓶中,用50mL 量筒各加30mL 二甲苯。
用100μL 微量注射器各加100μL 正十六烷标准溶液,再分别加入20μL、60μL、100μL、150μL、200μL、300μL 萘标准溶液,混匀,加盖保存备用。
②标准曲线的确定调整好色谱仪,用10μL 微量注射器分别抽取标样0.4μL,注入色谱仪。
测量正十六烷和萘的保留时间(s )和峰高(㎜),以保留时间与峰高的乘积作峰面积,或用积分仪直接测量正十六烷和萘的峰面积。
气相色谱法检测焦油中萘含量

图 3 柱温为一阶升温的色谱图
212样品的前处理方法称取煤焦油试样约05g称取0030g正十二烷精确到01mg可采用100l色谱注射器移取40l于带密封垫的样瓶中用移液管加入6mlnn二甲基乙酰胺在旋涡混合仪上振荡15秒将样品均匀气温较低时可采取先水浴加热再振荡用微量注射器取1l在规定的试验条件下进行分析
质量与检测
气相色谱法检测焦油中萘含量
叶敏(宝武集团鄂钢公司,湖北 鄂州 436000)
摘 要:通过对溶剂、内标物以及柱室温度的优化,建立了气相色谱-氢火焰光度检测器(GC-FPD)检测焦油中萘含量的方法。利 用所建立的方法对标样进行实验。结果表明,方法的准确度和精密度满足要求。 关键词:萘;焦油;气相色谱;内标法
经查阅有关资料[1-2], 煤焦油中萘含量测定方法有“蒸馏-结 晶点法”及行业标准“萃取-色谱(填充柱) 法”, 但该法分析时间 长, 操作步骤繁琐, 极易造成环境污染。用毛细管色谱法分析 煤焦油中萘含量的一种新方法, 为国内焦化行业煤焦油 加工工 艺的生产控制和产品质量检验提供了理想的方法依据。
HP-5 毛 细 管 柱 ,载 气 为 高 纯 氦 气(99.999%),流 速 2.635mL/min,检测器温度 300℃,进样口温度 300℃,氢气流速 35mL/min,空气流速 350mL/min,尾吹量 30mL/min,分流进样, 分流比为 50:1;进样量 1.0μL。 2.3 校正因子
在此实验条件下对焦化公司的焦油试样进行了分析(表 1), 结果表明,用气测数据
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
煤气中萘含量的测定
二、气相色谱法
1.方法原理、适用范围和引用标准
(1)方法原理用二甲苯或甲苯吸收煤气中的萘及其它杂质(茚、硫茚、甲基萘等),吸入液加入一定量内标液正十六烷,用气相色谱法分离,测定萘的含量。
(2)适用范围本标准规定了城市燃气中萘含量的气相色谱分析测定方法,适用于萘含量在5mg/m3以上的城市燃气。
(3)引用标准GB/T682《化学试剂三氯甲烷》;GB/T684《化学试剂甲苯》。
2.操作步骤
(1)调整仪器按下列条件调整仪器,允许根据实际情况作适当变动。
各组分的相对保留值见下表。
各组分的相对保留值
气相色谱条件如下:汽化温度,250℃;柱箱和色谱柱温度,恒温130℃;载气,氮气;柱前压,约73.5kPa(0.75kgf/cm2);流速,35mL/min(柱后测量);检测器,
火焰离子化检测器;检测器温度,140℃;辅助气流速度,氢气,40mL/min ,空气,400mL/min ;灵敏度和衰减的调节,在萘的绝对进样量为2.5×10-8g 时,产生的峰高不低于10mm ;记录仪纸速,1㎝/min 。
(2)校准
①标准样品的制备
正十六烷标准溶液:称取7.5g 正十六烷(称准至0.0002g ),置于50mL 容量瓶中,用二甲苯稀释至刻度,混匀,密封贮存备用,溶液浓度应定期检查。
萘标准溶液:称取7.5g 萘(称准至0.0002g ),置于50mL 容量瓶中,用二甲苯稀释至刻度,混匀,密封贮存备用。
校准用标准样品系列的制备:在6个50mL 的小口试剂瓶中,用50mL 量筒各加30mL 二甲苯。
用100μL 微量注射器各加100μL 正十六烷标准溶液,再分别加入20μL 、60μL 、100μL 、150μL 、200μL 、300μL 萘标准溶液,混匀,加盖保存备用。
②标准曲线的确定
调整好色谱仪,用10μL 微量注射器分别抽取标样0.4μL ,注入色谱仪。
测量正十六烷和萘的保留时间(s )和峰高(㎜),以保留时间与峰高的乘积作峰面积,或用积分仪直接测量正十六烷和萘的峰面积。
按下式分别计算各标准样品中萘和正十六烷的质量比Y i 和峰面积比X i 。
i
i i V V m m Y 2121⨯= i i i A A X 21=
式中 Y i —第i 个标准试样中萘与正十六烷的质量比;
m1—配制萘标准溶液时萘的称取量,g;
m2—配制正十六烷标准溶液时正十六烷的称取量,g;
V1i—配制第i个标准试样时所用萘标准溶液的体积,μL;
V2i—配制第i个标准试样时所用正十六烷标准溶液的体积,μL;
X i—第i个标准试样的萘与正十六烷的峰面积比;
A1i—第i个标准试样相应的萘的峰面积,以保留时间(s)与峰高(㎜)之乘
积表示或用积分仪测得的积分数表示;
A2i—第i个标准试样相应的正十六烷的峰面积,以保留时间(s)与峰高(㎜)
之乘积表示或用积分仪测得的积分数表示;
将X对Y作校准曲线,或用数学回归法建立如式(6-23)的线性回归方程:
Y=a+bX
注:每个标准试样进样三次,计算三次峰面积比后取算术平均值作图或进行数学回归。
(3)试验
①取样
准备工作:取样位置应避开煤气管道弯头或分叉处。
取样管为外径7㎜的不锈钢管,插入煤气主管的1/3处。
取样管直接与吸收瓶连接,其外露于煤气管外至吸收瓶的部分应尽量的短,并用热水夹套保温,使取样管中煤气的温度比煤气主管中煤气的温度高5~10℃。
两只各加30mL甲苯或二甲苯的吸收瓶置于加冰的冷水浴中,保证在取样时吸收液温度不高于10℃,在加热保温取样管后,置换放散煤气10min。
吸收:连接取样管、吸收瓶和湿式流量计。
取样管、吸收瓶之间的连接,使用橡胶
管或塑料管,管口应尽量互相对接,避免气样与连接管接触。
记下流量计读数,通入煤气,调节流速在0.5~1.0L/min 之内。
根据煤气中的萘含量,通入适量煤气,使被吸收的萘的总量在2~40mg 之间。
停止通气,记录吸收的煤气体积、煤气压力、温度及大气压,取下吸收瓶。
取样过程中,应注意避免吸收瓶入口处形成萘的结晶。
②吸收液的分析
在两个吸收瓶中,用100μL 微量注射器各加入100μL 正十六烷标准溶液,充分混匀,用洗耳球对吸收瓶的吸收管吹气,使吸收液置换数次,以保证混合均匀。
调整仪器的操作条件与进行标准试样分析时的条件相同。
用10μL 微量注射器抽取0.4μL 吸收液注入色谱仪进行分析。
测量正十六烷和萘的保留时间(s )与峰高(㎜),或用积分仪直接测量正十六烷和萘的峰面积。
每个吸收液各做两次分析。
第二个吸收瓶中所含萘应一并计算。
3.计算结果
按下式分别计算两吸收液中萘与正十六烷的峰面积比:
2
1A A X 式中 X —吸收液中萘与正十六烷的峰面积比;
A 1,A 2,—分别为萘和正十六烷的峰面积或积分仪的积分值。
根据X 从校准曲线上查出或用式(6-22)计算出Y 值,即为吸收液中萘与正十六烷的质量比。
按下式分别计算两种吸收液中的萘含量:
m=Ym s
式中 m —吸收液中萘的含量,mg ;
m s —加入吸收液中正十六烷的质量,mg ;
Y —吸收液中萘与正十六烷的质量比。
城市燃气中的萘含量按下式计算:
10000
⨯=V m c 式中 c —城市燃气中的萘含量,mg/m 3;
M —吸收液中萘的含量,mg ;
V 0—标准状态下干煤气的取样体积,L 。
V 0按下式计算:
()f p p p p t V V w g g -+⨯+⨯=0
10115.27315.273 式中 V 1—测定时流量计读取的气样体积,L ;
t g —测定时气样的温度,℃;
p 0—标准大气压的,等于101.325kPa (760㎜Hg );
p —测定时的大气压,Pa ;
p g —煤气压力,Pa ;
p w —测定温度下的饱和水蒸气压,Pa ;
f —流量计校正系数。