05多序列比对和进化树分析
05多序列比对和进化树分析

common carp
zebrafish
rainbow trout teleost
Orthologs: members of a gene (protein) family in various organisms. This tree shows RBP(视黄醇结合蛋白) orthologs.
Multiple sequence alignment programs How to get multiple sequences?
Sequence format BLAST Program
Multiple sequence alignment programs
Genedoc
Clustal X Clustal W Align X MultAlin T-Coffee MAFFT
Definitions: two types of homology Orthologs Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function.
2.采用ClustalW在线分析( AAQ84722.1 )
来的各分类单位间的相互关系。
离散特征法则主要包括 MP 法(最大简约法)和 ML 法(最大 似然法)。 距离法在构成距离矩阵(故而也称距离矩阵法)后,要么通过 某个标准来筛选出进化树的最佳估计,可以用最小二乘标准来 估计进化树,称最小二乘进化树;或者根据某种算法得到一个 聚类的树形图,不必对每个树都进行比较,计算量小,因此也 不一定是最佳的树,常见的有UPGMA法(类平均法)和NJ法 (neighbor-joining method,邻接法)。
生物信息学-第四章-多序列比对与分子进化分析

Clustal使用方法
Clustal:目前被最广泛应用的 MSA 方法
可在线分析
可在本地计算机运行 序列输入、输出格式
Input FASTA
NBRF/PIR EMBL/SWISSPROT ALN GCG/MSF GCG9/RSF GDE
>sequence 1 ATTGCAGTTCGCA … … >sequence 2 ATAGCACATCGCA… … >sequence 3 ATGCCACTCCGCC… …
10 3 2 5
C B
2
D
outgroup 外群、外围支
系统发育树构建步骤
多序列比对(自动比对、手工校正)
最大简约法 (maximum parsimony, MP) 距离法 选择建树方法(替代模型) (distance) 最大似然法 (maximum likelihood, ML) 贝叶斯法 (Bayesian inference) UPGMA
多序列比对的应用: •系统发育分析(phylogenetic analysis) •结构预测(structure prediction) •序列基序鉴定(sequence motif identification) •功能预测(function prediction) ClustalW/ClustalX:一种全局的多序列 比对程序,可以用来绘制亲缘树,分析进化 关系。 MEGA5——分子进化遗传分析软件
比对参数设置
两两比对参数
多序列比对参数
点击进行多序列比对
比对结果 “*”、“:”、“.” 和空格依次代表改位点的序列一致性由高到低
第四步:比对完成,选择结果文件的保存格式
可进一步对排列好的序列进行修饰(1)
mega操作过程-多序列比对、进化树、

基 在NCBI/EBI的FTP服务器上可以找到下载的软件包。
础 生
ClustalW 程序用选项单逐步指导用户进行操作,用户
物
可根据需要选择打分矩阵、设置空位罚分等。
信 息
ftp:///pub/software/
学
EBI的主页还提供了基于Web的ClustalW服务,用户可以
物
信
随着序列数量的增加,算法复杂性也不断增加。用O
息
(m1m2m3…mn)表示对n个序列进行比对时的算法复杂性,
学
其中mn是最后一条序列的长度。若序列长度相差不大,则
及 应
可简化成O(mn),其中n表示序列的数目,m表示序列的长
用
度。显然,随着序列数量的增加,序列比对的算法复杂性
按指数规律增长。
第二节 多序列比对程序及应用
及 应
把序列和各种要求通过表单提交到服务器上,服务器
用
把计算的结果用Email返回用户(或在线交互使用)。
/clustalw/
Progressive Alignment Method
ClustalW 程序
基
ClustalW对输入序列的格式比较灵活,可以是FASTA格式,还可
1 2 3 4 5 6 7 8 91
ⅠY D G G A V - E AL
基
础
ⅡY D G G - - - E AL
生
物
ⅢF E G G I L V E AL
信
息
学
ⅣF D - G I L V Q AV
及
应
ⅤY E G G A V V Q AL
用
表1 多序列比对的定义
表示五个短序列(I-V)的比对结果。通过插入空位,使5个序列中 大多数相同或相似残基放入同一列,并保持每个序列残基顺序不变
mega操作过程-多序列比对、进化树、

用ClustalW得到的多序列比对结果中,所有序列排列在一起,并
以特定的符号代表各个位点上残基的保守性,“*”号表示保守性 极高的残基位点;“.”号代表保守性略低的残基位点。
Progressive Alignment Method
Clustal W 使用
输入地址: 设置选项 (next)
用于描述一组同源序列之间的亲缘关系的远近,应用到 分子进化分析中。 序列同源性分析:是将待研究序列加入到一组与之 同源,但来自不同物种的序列中进行多序列同时比 较,以确定该序列与其它序列间的同源性大小。
其他应用,如构建profile,打分矩阵等
3、多序列比对的方法
手工比对 在运行经过测试并具有比较高的可信度的计算机程序(辅助 编辑软件如bioedit,seaview,Genedoc等)基础上,结合实 验结果或文献资料,对多序列比对结果进行手工修饰,应该 说是非常必要的。 为了便于进行交互式手工比对,通常使用不同颜色表示具有 不同特性的残基,以帮助判别序列之间的相似性。
Extremely slow computation.
Progressive Alignment Method
DbClustal: Poa (Partial order alignments):
2、Iterative Alignment
PRRN:
web-based program Nhomakorabea/
Uses a double nested iterative strategy for multiple alignment.
DCA (Divide-and-Conquer Alignment):a web-based program that is semiexhaustive /
序列比对,构建进化树
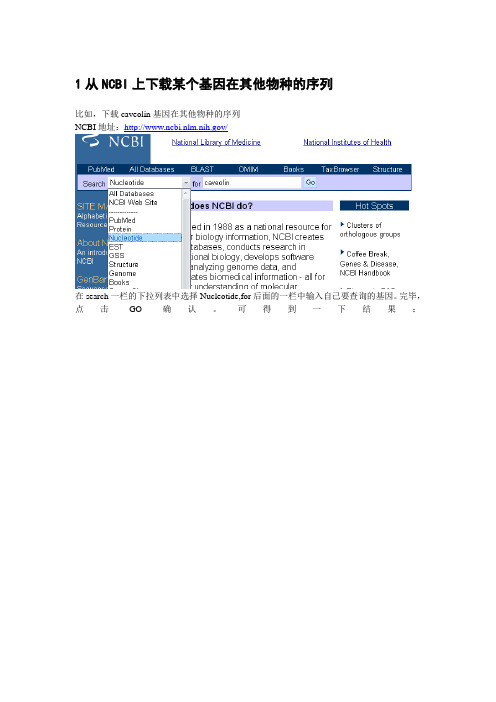
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
课件第4讲_多序列比对和进化分析

渐进法的策略I.将序列两两比对II.根据相似值将序列分组III.进行组间比对,并继续分组,直至取得最终结果Principle:比对过程中,相似性高的序列先比对,距离远的序列添加其后值与分歧时间t呈非线性关系,原因之一:多个氨基酸替代出现在同一位点。
基于泊松分布对p进行校正,得两序列间每位paralogsorthologs paralogs orthologsErik L.L. Sonnhammer Orthology,paralogy and proposedand proposed classification for paralog subtypes TRENDS in Genetics Vol.18 No.12 December 2002UPGMA方法例:OTU1和OTU2都是原始类群,n1=1,n2=1 OTU r1含两个原始类群OTU1和OTU2 ,nr1=2,OTU3是原始类群,n3=1简明生物信息学,钟扬等主编,用UPGMA法构建的系统树常用构树法比较/phylip/s oftware.htmlHere are 386phylogeny packages and 52free servers, all that I know about. It is an attempt to be completely comprehensive. I have not made any attempt to exclude programs that do not meet some standard of quality or importance….Many of the programs in these pages are available on the web, and some of the older ones are also available from ftp server machines.。
生物信息学 第五章 多序列比对

多序列比对有时用来区分一组序列之间的差异,但其主要用于描述一组序列之间的相 似性关系,以便对一个基因家族的特征有一个简明扼要的了解。与双序列比对一样,多序列 比对的方法建立在某个数学或生物学模型之上。因此,正如我们不能对双序列比对的结果得 出“正确或错误”的简单结论一样,多序列比对的结果也没有绝对正确和绝对错误之分,而 只能认为所使用的模型在多大程度上反映了序列之间的相似性关系以及它们的生物学特征。 显然,多序列比对需要使用许多专门的分析工具。除了一些已经广泛使用并仍在不但 改进的多序列计算机程序外,还需要有一个开发方便实用的多序列比对手工编辑工具。 可以从多个不同角度出发构建多序列比对模型。这里,主要指建立比对模型的生物学 基础,而不仅是具体的比对方法,如自动比对或手动比对等。目前,构建多序列比对模型的 方法大体可以分为两大类。第一类是基于氨基酸残基的相似性,如物化性质、残基之间的可 突变性等。另一类方法则主要利用蛋白质分子的二级结构和三级结构信息,也就是说根据序 列的高级结构特征确定比对结果。显然,这两种方法所得结果可能有很大差别。一般说来, 很难断定哪种方法所得结果一定正确,应该说,它们从不同角度反映蛋白质序列中所包含的 生物学信息。 基于序列信息和基于结构信息的比对都是非常重要的比对模型,但它们都有不可避免 的局限性,因为这两种方法都不能完全反映蛋白质分子所携带的全部信息。我们知道,蛋白 质序列是经过 DNA 序列转录翻译得到的。从信息论的角度看,它应该与 DNA 分子所携带 的信息更为“接近”。而蛋白质结构除了序列本身带来的信息外,还包括经过翻译后加工修 饰所增加的结构信息,包括残基的修饰,分子间的相互作用等,最终形成稳定的天然蛋白质 结构。因此,这也是对完全基于序列数据比对方法批评的主要原因。显然,如果能够利用结 构数据,对于序列比对无疑有很大帮助。不幸的是,与大量的序列数据相比,实验测得的蛋 白质三维结构数据实在少得可怜。在大多数情况下,并没有结构数据可以利用,我们只能依 靠序列的相似性和一些生物化学特性建立一个比较满意的多序列比对模型。
生物信息学中的序列比对与序列分析研究

生物信息学中的序列比对与序列分析研究序列比对与序列分析是生物信息学领域中非常重要的研究内容之一。
在基因组学和蛋白质组学的快速发展下,对生物序列的比对和分析需求不断增长。
本文将介绍序列比对和序列分析的概念、方法和应用,并探讨其在生物学研究中的重要性。
一、序列比对的概念与方法:1. 序列比对的概念:序列比对是将两个或多个生物序列进行对比,确定它们之间的相似性和差异性的过程。
在生物信息学中,序列通常是DNA、RNA或蛋白质的一连串碱基或氨基酸。
序列比对可以用来寻找相似性,例如发现新的基因家族、识别保守的结构域或区分不同的物种。
2. 序列比对的方法:序列比对的方法可以分为两大类:全局比对和局部比对。
全局比对将整个序列进行比对,用于高度相似的序列。
而局部比对则将两个序列的某个片段进行比对,用于相对较低的相似性。
最常用的序列比对算法是Smith-Waterman算法和Needleman-Wunsch算法。
Smith-Waterman算法是一种动态规划算法,它在考虑不同区域的匹配得分时,考虑到了负分数,适用于寻找局部相似性。
而Needleman-Wunsch算法是一种全局比对算法,通过动态规划计算最佳匹配得分和最佳比对方式。
二、序列比对在生物学研究中的应用:1. 基因组比对:序列比对在基因组学中具有广泛的应用。
它可以帮助研究人员对特定基因进行鉴定,发现重要的调控元件以及揭示物种间的基因结构和功能差异。
此外,基因组比对还可以用于揭示突变引起的遗传疾病和肿瘤等疾病的发病机制。
2. 蛋白质结构预测:序列比对在蛋白质结构预测中也起着重要的作用。
通过将待预测蛋白质序列与已知结构的蛋白质序列进行比对,可以预测其二级和三级结构以及可能的功能区域。
这些预测结果对于理解蛋白质的功能和相互作用至关重要。
3. 分子进化分析:序列比对在分子进化研究中也扮演着重要的角色。
通过将源自不同物种的基因或蛋白质序列进行比对,可以构建进化树,研究物种的亲缘关系和演化历史。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
http://tcoffee.crg.cat/apps/tcoffee/do:regular
多序列比对软件——MAFFT
rpm –ivh mafft-7.305-gcc_fc6.x86_64.rpm 必须有root权限
Download and installation
多序列比对软件——MAFFT
Align X (1) 序列的输入
(2) 序列alignment
(3) 结果的编辑( Metafile; text )
Multalin
http://multalin.toulouse.inra.fr/multalin/
T-Coffee
Multiple Sequence Alignment Tools
Definitions: two types of homology Orthologs Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function.
运行步骤
(1) (2) (3) 序列的输入,1,输入序列的名称 序列alignment 选择,2,1,或者其他选项 运行,
(4)
结果导出
进到下面这个文件夹 cd src/ 运行即可 ./clustalw2
Clustal W
Bioedit
/clustalv
Software
1. ClustalX +Treeview 2. Mega 3.1
/mega.html
进化树的应用
1. 新基因的鉴定 2. 新蛋白的分类
蛋白质功能预测
1. 同源蛋白功能推测; 2. 蛋白质结构域或基元分析。
Pattern and profile searches
利用现代分子生物学技术所获得的生物多样性的信息 ,可大致
分为以下两大类:1)离散特征数据 (discrete character data),
即所获得的是 2个或更多的离散的值 ,是赋给某一具体的运筹 分类单位(operational taxonomic unit ,简称OTU)的;2)相 似性和距离数据 (similarity and distance data),它并不是某一 具体分类单元所具有 , 而是用彼此间的相似性或距离所表示出
Sequence alignment of S_TKc domain of PXK_v1 with consensus S_TKc domain. Identical residues are represented in black and similar residues in gray. The subdomains of the S_TKc domain are indicated with Roman numerals. Asterisks denote the indispensable residues of lysine, glutamine and aspartic acid in consensus S_TKc domain.
Sequence alignment of Homo sapiens Sgt1.2 with its five homologous proteins. Numbers on the right refer to the last amino acid in each corresponding line. Residues indicated with dark shading are identical amino acids. Grey shading represents 80-90% similarity and light grey means 60-70% similarity.
生物信息学
第五章 多序列比对和进化树分析
Part I
Sequence alignment
Definitions
Pairwise alignment The process of lining up two or more sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology.
Genedoc
(1) 序列的输入 (2) 序列alignment
(3) 格式调节
(4) 输出到绘图内编辑
Alignment of A. ferrooxidans SOD protein and its orthologs. Atf27230: A. ferrooxidans ATCC 27230, De195: Dehalococcoides ethenogenes 195 Gspca: Geobacter sulfurreducens PCA, Tad1728: Thermoplasma acidophilum DSM 1728. Identical residues have been boxed and are shaded in dark.
African clawed frog chicken human horse pig cow 10 changes rabbit
mouse rat
apolipoprotein D retinol-binding protein 4 Complement component 8 Alpha-1 Microglobulin /bikunin
Multiple sequence alignment programs How to get multiple sequences?
Sequence format BLAST Program
Multiple sequence alignment programs
Genedoc
Clustal X Clustal W Align X MultAlin T-Coffee MAFFT
Paralogs Homologous sequences within a single species that arose by gene duplication.
common carp
zebrafish
rainbow trout teleost
Orthologs: members of a gene (protein) family in various organisms. This tree shows RBP(视黄醇结合蛋白) orthologs.
2.采用ClustalW在线分析( AAQ84722.1 )
Paralogs: members of a gene (protein) family within a species
prostaglandin D2 synthase progestagenassociated endometrial protein neutrophil gelatinaseassociated lipocalin
Clustal X
(1) 序列的输入 (2) 序列alignment
Clustal W
ClustalW(命令行)是ClustalX(图形版)的姊妹版,在DOS或linux下运行 安装:
首先解压压缩包 tar -xzvf clustalw-2.1.tar.gz 进到解压后的文件夹 cd clustalw-2.1 安装 ./configure make
Definitions
Homology Similarity attributed to descent from a common ancestor.
Identity The extent to which two (nucleotide or amino acid) sequences are invariant. Similarity The extent to which two (nucleotide or amino acid) sequences are similar.
Odorant-binding protein 2A
Lipocalin 1
10 changes
How to calculate similarity and identity?
1. Align X 2. MatGAT 3. Bioedit
Align X
Align X is one of the standalone of Vector NTI suite Not easy to get the cracked version
SMART
http://smart.embl-heidelberg.de/smart/set_mode.cgi?NORMAL=1
InterProScan
Motifscan
作业
1.采用Genedoc软件分析( AAQ84722.1 )
要求:4个ortholog蛋白质序列alignment,每 排 80个氨基酸残基,采用二色(黑色标记一 致氨基酸残基),每一个比较的蛋白质给出 Genbank登录号
多序列比对文件美化
GeneDoc Boxshade Espript TEXshade WebLogo/SeqLogo JProfileGrid
多序列比对结果特征提取
Protein alignment based DNA alignment
http://www.cbs.dtu.dk/services/RevTrans/