生物信息学-第五章-核苷酸序列分析
生物信息学核酸序列分析

此外,运行 Motif 程序可在共有序列中搜索已知的蛋白质模式。Motif 在蛋白质序列中 搜索在 PROSITE,蛋白质位点和模式的 PROSITE 字典中已知的蛋白质模式。如果辨识出一个 Motif,则给所有序列增加一个特征,并标出它的位置。图 4.9 显示了一个蛋白质序列的匹 配、一个共有序列以及 Motif 搜索的结果。
(3)用查询序列搜索数据库,将找到的条目与查询序列进行对比并产生进化系统树
克隆并测序一个未知功能基因的用户可能希望在一个数据库中搜索相似的序列。如果搜 索到了,用户可能进一步希望创建与查询序列最相似的序列的多序列对比并产生数据的种系 图。
往 SeqLab Editor 中添加一个查询序列并从 Functions 菜单中选取 FASTA 程序。FASTA 程序在数据库中搜索与查询序列相似的序列。输出文件可从 Output Manager 窗口中加以显 示并直接添加到 SeqLab Editor 中。在这个输出文件中数据库条目与查询序列局部相似性最 好的区域被加以标记。如果要显示的话,每个数据库条目只有这种区域可以显示在 SeqLab Editor 中。不要的条目可以从 SeqLab Editor 中一起被删除。
(5)对比相关的蛋白质序列,计算对比结果的共有序列,辨识序列中新的特征序列模式,在 数据库中搜索包含此模式的序列或在对比结果的共有序列中搜索已知的蛋白质模式
辨识了一组相关序列的用户可能希望对其进行对比并计算对比结果的共有序列。如果可 以在对比结果中找到保守模式,用户可能希望在数据库中搜索包含这种模式的其它序列。用 户可能还希望在计算出的共有序列搜索已知的蛋白质模式。
核苷酸序列分析
核苷酸序列分析
ORF
Getorf
Plotorf ORF Finder BestORF
基因开放阅读框/基因结构分析识别工具
http://bioweb.pasteur.fr/seqanal/interfaces/getorf.html
http://bioweb.pasteur.fr/seqanal/interfaces/plotorf.html /gorf/gorf.html /all.htm
• GlimerM适于恶性疟原虫、拟南芥、曲霉菌 和水稻 • 对mRNA, cDNA, EST, 宜用GetOrf, ORF Finder, Plotorf, BestORF 等
核苷酸序列分析
ORF
应用ORF Finder预测水稻瘤矮病毒 (RGDV)S8片断的ORF
• ORF Finder: /gorf/gorf.html
核苷酸序列分析
重复序列分析 开放读码框(open reading frame, ORF)的识别 基因结构分析 内含子/外显子剪切位点识别 选择性剪切分析 CpG 岛的识别 核心启动子/转录因子结合位点/转录启始位 点的识别 转录终止信号的预测 GC含量/密码子偏好性分析
核苷酸序列分析
ORF
重复序列分析
Web/Linux
Web Web Web/Linux Linur
FGENESH+ /++
/generation/
r.it/~webgene/genebuilder.html /all.htm /genomescan.html /Software/Wise2/ /grailexp/ /seq-search/genesearch.html
• Kozak规则: ORF中起始密码子ATG前后的碱基具 有特定的偏好性。若将第一个ATG中的碱基分别 标为1、2、3位,则Kozak规则可描述如下:
核苷酸序列物种鉴定

核苷酸序列物种鉴定全文共四篇示例,供读者参考第一篇示例:核苷酸序列物种鉴定是一种通过对生物体DNA或RNA序列进行分析,从而确定生物物种归属的技朧。
随着生物技术的发展,核苷酸序列物种鉴定已经成为一种常用的方法,被广泛应用于生物学、生态学、医学等领域。
本文将介绍核苷酸序列物种鉴定的基本原理、方法以及在不同领域的应用。
一、核苷酸序列物种鉴定的基本原理核苷酸序列物种鉴定的基本原理是利用生物体DNA或RNA的特定序列来确定其物种归属。
每个生物种类都有自己独特的核苷酸序列,这些序列通常被称为“基因组指纹”。
基因组指纹在所有个体中都是唯一的,可以用来区分不同的物种。
核苷酸序列物种鉴定通常包括以下几个步骤:提取生物体DNA或RNA,对其进行PCR扩增,测序获得核苷酸序列,与数据库中已知物种的核苷酸序列比对,最终确定物种。
1. 提取DNA或RNA:首先需要从生物体中提取DNA或RNA。
通常使用的提取方法包括CTAB法、蛋白酶K法等。
2. PCR扩增:利用PCR技术对提取的DNA进行扩增,通常选择一些具有物种特异性的基因作为扩增靶标。
常用的扩增靶标包括rDNA、COI等。
3. 测序:对PCR扩增产物进行测序,获取核苷酸序列。
三、核苷酸序列物种鉴定在生物学、生态学、医学等领域的应用1. 生物学领域:在生物多样性研究中,核苷酸序列物种鉴定可以用来确定生物物种的归属,推测物种进化关系,研究物种分布、种群结构等。
3. 医学领域:在医学诊断中,核苷酸序列物种鉴定可以用来快速检测病原微生物,帮助医生确定病原体种类,指导治疗方案。
核苷酸序列物种鉴定具有很高的精准度和灵敏度,是一种非常有效的物种鉴定方法。
随着生物技术的不断发展,相信核苷酸序列物种鉴定在未来将会在更多领域得到广泛应用,为人类的生活和科学研究提供更多便利。
【2000字】第二篇示例:核酸序列是生物体中含有遗传信息的一种序列。
对于不同的生物种类,其核酸序列会呈现出不同的特征,这使得核酸序列成为一种用于物种鉴定的重要工具。
生物信息学 第五章 核酸序列分析

第五章 核酸序列分析
生物科学与技术学院
▪ ▪ 不同基因组中两个连续核苷酸出现的频率也是不相同的 4种核苷酸可以组合成16种两联核苷酸
酵母基因组两联核苷酸频率表
设:Pij代表两联核苷酸(i,j)的出现频率;Pi 代表核苷酸i的出现频率 则:Sij= Pij/(PiPj), Sij反应了核苷酸i和j的 关联关系,若Sij=1,则在两个连续的位 置上,核苷酸i和j的出现是相对独立的。 若Sij>1,则两个连续位置上,核苷酸i 和j的出现是相关的。 如:酵母基因组P(A)=0.3248,
Codon Usage Analyzer
/codon/cgi-bin/codon.cgi
三、GC含量分析
GC含量 (GC content):是基因组的基本参数,即DNA分子或基因组中GC碱基
对所占的比例,通常用百分比表示,如15~75%。物种的GC含量存在两头少中间 多的正态分布情况。GC含量可用分光计测量,DNA的解链温度(解链时260nm光
AA和AT、TCG、ATC、GCA、A。这三种顺序被称为开放阅读框。
实现方法: ① 扫描给定的DNA序列,在3个不同的阅读框中寻找较长的ORF。
② 当遇到终止密码子后,回头寻找起始密码子,以确定完整的编码区域。
基因开放阅读框/基因结构分析识别工具
Getorf Plotorf ORF Finder BestORF GENSCAN Gene Finder FGENESH GeneMark http://bioweb.pasteur.fr/seqanal/interfaces/getorf.html http://bioweb.pasteur.fr/seqanal/interfaces/plotorf.html /gorf/gorf.html /all.htm /GENSCAN.html /tools/genefinder/ /all.htm /GeneMark/ EMBOSS EMBOSS NCBI Softberry MIT Zhang lab Softberry GIT 通用 通用 通用 真核 脊椎、拟南芥、玉米 人、小鼠、拟南芥、酵母 真核 原核
生物信息学中的DNA和RNA序列分析方法

生物信息学中的DNA和RNA序列分析方法随着生物研究的发展,生物信息学逐渐成为了一个十分重要的学科领域,DNA和RNA序列分析是其中较为重要的一个方面。
DNA和RNA是生物体中的核酸,它们携带了生命的遗传信息,而对这些信息进行解读和分析就需要运用到生物信息学。
本文将为大家介绍生物信息学中的DNA和RNA序列分析方法。
一、基础知识在深入了解DNA和RNA序列分析方法之前,我们需要先了解一些基础知识。
1. DNA和RNA的基本结构DNA双链螺旋结构由核苷酸组成,其中核苷酸由磷酸、五碳糖核糖或脱氧核糖和一种氮碱基组成。
常见的氮碱基有腺嘌呤(A)、鸟嘌呤(G)、胸腺嘧啶(T)和胞嘧啶(C)。
RNA是由核苷酸组成的单链分子,比DNA少了胸腺嘧啶,而是由尿嘧啶(U)取代了。
2. DNA和RNA的编码DNA编码了基因信息,而RNA通过转录形成mRNA,再到翻译形成蛋白质。
在转录过程中,mRNA中的氮碱基按照特定的规则与DNA上的氮碱基匹配,即腺嘌呤与尿嘧啶配对,鸟嘌呤与胞嘧啶配对。
这种配对方式被称之为互补配对。
RNA与DNA的互补配对非常重要,因为它决定了RNA能够识别和复制DNA中的信息。
二、DNA和RNA序列分析方法DNA和RNA序列分析方法主要有以下几种。
1. 序列比对序列比对是指将两个或多个序列进行比较,找出它们之间的相似处和差异。
序列比对是进行生物信息学研究的基础,也是DNA 和RNA序列分析的核心方法。
序列比对有两种类型,全局比对和局部比对。
全局比对一般用来比较两个完整的序列,例如蛋白质序列。
局部比对一般用来比较一个序列中的一小段与另一个序列中的一小段。
2. 序列注释序列注释是指将序列上的功能信息注释到序列上。
一般情况下,序列注释会包括以下几个方面的信息:基因结构,包括外显子、内含子、UTR等;转录因子结合位点、启动子和增强子等调控元件;蛋白质结构,包括功能和结构域等;翻译起始和终止位点等。
序列注释需要利用已知的信息,例如已知的基因、蛋白质和调控元件等数据库信息。
核苷酸序列
核苷酸序列分析 ORF
启动子及转录因子结合位点分析
• 启动子(Promoter)是RNA聚合酶识别、结合并开 始转录所必需的一段DNA序列。
• 原核生物启动子序列包括:
1. CAP序列(增强聚合酶的结合和转录的起始序列,70~-40)
2. -10序列:在-4到-13bp处,有保守序列TATAAT,称为 Pribnow框,各碱基频率:T89 A89 T50 A65 A65 T100
3. -35序列:约在-35处有保守序列TTGACA, 其中TTG十 分保守,各碱基频率:T85 T83 G81 A61 C69 A52
核苷酸序列分析 ORF
启动子及转录因子结合位点分析
• 真核生物启动子是在基因转录起始位点(+1)及其5’ 上游大约100~200bp或下游100bp的一组具有独立 功能的DNA序列,包括:
核苷酸序列分析 ORF
重复序列分析
2. 中度重复序列。长10~300bp,重复10~105次, 占基因组10~40%。哺乳类中含量最多的一种 称为Alu的序列,长约300bp,重复3×105次, 在人类基因组中约占7%,功能不是很清楚。
3. 单拷贝序列。这类序列基本上不重复,占哺乳 类基因组的50%~80%,在人类基因组中约占 65%。
输出结果
GENSCAN
ggccagatgg aacatattgc tttcgggagc acaaggatcg ggtctactac gtctcggagc ggattttgaa gctgagcgag tgcttcggct acaagcagct ggtgtgcgtg ggcacctgct tcggcaagtt ctccaagacc aacaaactga agttccatat cacggcgctc tactacttgg cgccctacgc ccagtacaag gtgtgggtga agccctcctt cgagcagcag tttctctacg
实验一生物信息学资源的利用—核苷酸序列的查找
实验一生物信息学资源的利用—Genebank核苷酸序列的查找一、实验目的:了解生物信息学的各大门户网站以及其中的主要资源,并以NCBI提供的Genebank为例,学习蛋白质及核苷酸序列的检索方法和使用技巧。
二、实验器材:计算机,NCBI、EMBL等生物信息学网络资源。
三、实验原理:根据Genebank 提供的数据资源,应用分类学方法进行核苷酸序列的查找。
四、实验内容:查找下列不同物种的NAC家族的核苷酸序列及蛋白序列。
(Arabidopsis thaliana; Oryza sativa; Zea mays; Solanum lycopersicum)每小组找五条序列。
五、实验步骤:1、打开NCBI网站的主页,搜索栏中输入“NAC”然后点击Search,2、选择蛋白序列数据库(Protein: sequence database)进入到搜索结果界面,3、点击右则“RefSeq”(去冗余结果),在“Top Organisms”选项卡下,选择所需要的物种,单击一条记录,分别下载序列介绍和序列文件(fasta格式)。
4、找到蛋白对应的核酸序列,并下载。
六、实验要求:每个小组必须至少查找1个种,5条序列(十条蛋白序列及对应的核酸序列)。
必须写明查找到的序列以及各条序列的GenBank收录号-LOCUS,基因注释-DEFINITION,文章的作者AUTHORS,文章题目-TITLE,文章所发表的期刊-JOURNAL。
将序列文件单独保存至fasta格式。
七、实验结果:查找的核苷酸序列基本情况表1LOCUS NP_001078343 423 aa linear PLN 28-MAY-2011 DEFINITION NAC domain-containing protein 68 [Arabidopsis thaliana].AUTHORS Mayer,K., Schuller,C., Wambutt,R., Murphy,G., Volckaert,G.,TITLE Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana JOURNAL Nature 402 (6763), 769-777 (1999)实验二序列比对软件—BLAST和Clustal的使用一、实验目的:掌握序列相似性查询工具—BLAST(网络版和本地版)使用方法和技巧,理解与序列相似性查询相关的几个基本概念。
第五节核酸的序列分析
即核酸DNA分子一级结构的测定,是现代分子生 物学一项重要的技术。 1963年,Sanger和Thompson等人第一次完成 胰岛素51个氨基酸的序列测定,这在当时来说是 一件了不起的大事。70年代后期,Sanger和 Maxam----Gilbert等人又建立了核酸序列测定的 方法,Sanger双脱氧末端终止法和Maxam---Gilbert化学裂解法将核酸序列测定技术推进到 “直读”阶段,使核酸序列测定变得远比蛋白质 氨基酸序列测定容易,这样人们可以通过核酸序 列和遗传密码推导出蛋白质氨基酸的序列。
碱基特异性修饰及裂解
反应体系 碱基修饰试剂 碱基修饰反应 主链断裂试剂 G 硫酸二甲酯 鸟嘌呤甲基化 六氢吡啶 断裂点 G
G+A
C+T C
甲酸
肼 肼(加盐)
脱嘌呤作用
嘧啶开环 胞嘧啶开环
六氢吡啶
六氢吡啶 六氢吡啶
G和A
C和T C
化学法读序
每组测序图谱为4条垂直的阶梯带,该片从胶底部一
个个向顶部读,G+A和C+T两列中含有所有相差一个碱基的
目前市场上已经有各种型号的 DNA自动测序仪可供选购。
“分子生物学研究方法”思考题:
1、常用的核酸的凝胶电泳是哪两种?它们分辨 DNA片段的范围有何不同? 2、电泳操作的基本程序有哪些? 3、核酸的分子杂交的概念? Southern 印迹法、 Northern 印迹法和Western 印迹法的检测对象 分别是什么? 4、什么叫探针(Probe)? 5、PCR定义?简述聚合酶链式反应的基本原理? 6、核酸测序的常用方法有哪些?
优点
简便、迅速、应 用广泛。
不需酶促反应,可 1、高负荷,1块胶可 以对寡核苷酸测序。 测16个样品;2、机 读不需放射自显影; 3、安全不用同位素; 4、简单迅速。
核酸序列分析ppt课件
第一节 核酸序列的检索
一、 Entrez检索系统
(/sites/gquery?itool=toolbar)
二、 SRS 检索系统
()
三、DBGET/LinkDB检索
第二节 核酸序列的基本分析
一、 分子质量、碱基组成、碱基分布
/unigene
二、基因的电子定位分析
通过序列标签位点(STS)定位 通过UniGene/RH技术定位 利用基因组序列定位
1. 利用STS数据库进行定位
利用NCBI的电子PCR资源
(/sutils/e-pcr/forward.cgi)
()
四、克隆测序的分析
1. 测序峰图的查看
澳大利亚Conor McCarthy开发的Chromas.exe程序, 且BioEdit软件和DNAMAN软件都可以查看。
2. 核酸测序载体序列的识别与去除
测序克隆被宿主菌核酸序列污染,或目的克隆 来自于宿主菌,可通过Blastn直接对GenBank或 EMBL数据库进行相似性分析进行判断。
核酸序列分析
核酸序列分析是生物信息学应用中的一个重 要方面,一般包括:DNA碱基组成、密码子的偏 向、内部重复序列、特殊位点(限制性位点及转 录、翻译和表达调控相关信号)、编码区分析、 一二级结构等。
第一节 核酸序列的检索 第二节 核酸序列的基本分析 第三节 核酸序列的电子延伸 第四节 基因的电子表达、定位分析 第五节 基因识别 第六节 核酸序列的提交
终止密码子(TGA、TAA或TAG)数量较少; ORF达到一定的长度; 密码子使用的偏好性,第3个碱基G/C出现的频率较高; 与已知基因比较有序列相似性; 与模板序列的模式相匹配可能指示功能性位点的位置。
编码区的一些信号:
基因工程(基因工程的主要技术与原理-核苷酸序列分析)课件
核心原理:
利用特定的化学试剂对不同碱基进行特异 性切割。
硫酸二甲酯: 哌啶甲酸: 肼+NaCl: 肼:
G G和A C T和C
基因工程(基因工程的主要技术与原理核苷酸序列分析)
5′ 3′
G A+G
3′ 5′
待测DNA
放射性标记5′末端 R
限制性酶切
基因工程(基因工程的主要技术与原理核苷酸序列分析)
(二) 序列分析的基本步骤
模板变性(dnature template):将待测DNA模板 与引物混合,通过加热使模板变性; 退火(annealing):将变性的模板与引物混合物 缓慢降温,使引物与模板结合;
3. 分离:通过凝胶电泳分离片段群;
4. 推导:再经放射线自显影,确定各片段末端碱基, 从而得出目的DNA的碱基序列。
基因工程(基因工程的主要技术与原理核苷酸序列分析)
凝胶电泳分离,放射线ቤተ መጻሕፍቲ ባይዱ显影分析
G A+G C+T C 3′
5′ 5′ C T T基因T工T程(基T因T工程G的G主要G技术C与原T理T- A G C 3′
通过凝胶电泳分离,放射自显影确定DNA片段 末端的碱基,进而推断DNA的核苷酸序列。
基因工程(基因工程的主要技术与原理核苷酸序列分析)
5´ 3´
5´ 3´
5´ 3´
正常的DNA合成反应基因工程(基因工程d的dN主T要P技掺术与入原到理-DNA合成反应后导致反应终止
核苷酸序列分析)
基于双脱氧核苷酸的这种特性,Sanger于 1977年建立了以双脱氧链终止反应为基础来 测定DNA序列的方法;
该方法以待测DNA为模板,在DNA聚合酶的 催化作用下合成新的DNA链;
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
预测工具:
GENSCAN,GENEMARK NetGene2, Splice View
基因结构分析
内含子/外显子剪切位点识别
如何分析mRNA/cDNA的外显子组成?
RNASPL(软件) 与相应的基因组序列比对,分析比对片段的 分布位置 预测工具:
Spidey,SIM4,BLAT,BLAST,FASTA
FgeneSB
Softberry
细菌
FgeneSV
Generation FGENESH+ GenomeScan
/all.htm
/generation/ /all.htm /genomescan.html
选择性剪接是调控基因表达的重要机制 了解不同物种、细胞、发育阶段、环境压力下基因 的调控表达机制
分析方法: 查询选择性剪切相关的网站 多序列比对
基因结构分析
查询选择性剪切相关的网站
从已知基因的功能推测剪切机制
/asd/index.html http://splicenest.molgen.mpg.de/ /new_alt_exon_db2/
Softberry
ORNL Softberry MIT
病毒
原核 原核 脊椎、拟南芥、玉米
GeneWise
GRAIL
/Wise2/
/grailexp/
EBI
ORNL
人、蠕虫
人、小鼠、拟南芥、果蝇
基因预测
选择物种
可同时输入多条cDNA/mRNA序列与同一条基因组序列进行分析
输入基因组序列 或序列数据库号
判断用于分析的序列间的差异, 并调整比对参数 比对阈值 选择物种
输入mRNA.txt文档中的 6条序列
不受默认内含子长度限制, 默认长度:内部内含子 为35kb, 末端内含子为100kb 输出格式
基因结构分析
预测外显子位置、 可信度等信息
同源 比对 信息
基因预测
GenomeScan输出结果:图形
基因结构分析
基因结构分析
内含子/外显子剪切位点识别
对基因组序列的读码框区域进行预测
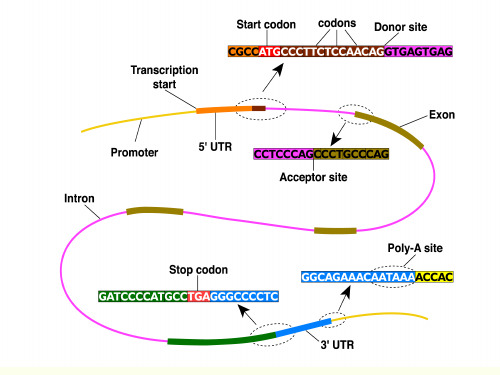
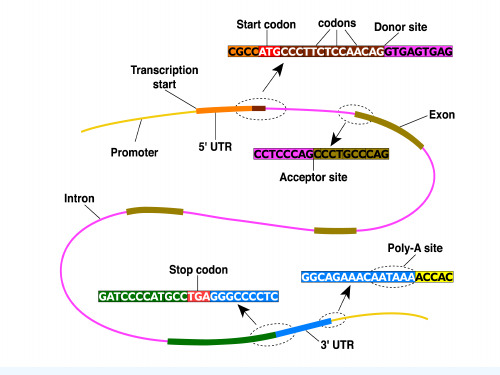
内含子5’端供体位点(donor splice site): GT 内含子3’端受体位点(acceptor splice site): AG
CpG岛 转录终止信号 GC含量
转录调控序列 分析
序列组分分析
限制性核酸内切酶位点 密码子偏好性使用
开放读码框的识别
• 开放读码框(open reading frame, ORF) 是一段起始密码子和终止密码子之间的碱基序列 • ORF 是潜在的蛋白质编码区
What does this sequence mean?
核苷酸序列分析
GENSCAN
基因预测
开放读码框
GenomeScan GLIMMER NetGene2 Spidey ProSplicer Spidey EPD Cister CpGPlot Hcpolya genskew NEBcutter CodonW
内含子/外显子剪切位点
基因结构分析
选择性剪切 启动子/转录起始位点
/all.htm /tdb/GeneSplicer/gene_spl.html /cgi-bin/sp.cgi
Web
Web Web/Linux Web
1 输入GI号或Accession,或直接输 NCBI ORF finder 入序列的 fasta 格式
2 结果出现六个图形,这是根据六种不同的 编码方式得到的(包括正反链)。
• 3 拿到氨基酸序列后,你可以直接做blastp, 如果有匹配到,就是正确的ORF区了。另外也 可以用Pfam的方法,在Pfam数据库搜索。
第三章 核苷酸序列分析
基因组功能分析
基因组序列 cDNA序列
翻译
编码区预测
蛋白质序列
蛋白质理化性质 二级结构预测 结构域分析 重要信号位点分析 三级结构预测
基因结构分析
序列比对 功能注释 KEGG GO 系统发育树 Codon bias 选择性剪切 GC Content 转录调控因子 限制性酶切位点
Web/Windows/ Linux
Web/Windows/ Linux
基因结构分析
剪切位点识别:NetGene2
http://www.cbs.dtu.dk/services/NetGene2/
基因结构分析
NetGene2输出结果
供体位点 可信度
受体位点
基因结构分析
mRNA剪切位点识别:Spidey
基因结构分析
选择性剪切数据库:ProSplicer
.tw/
基因名、数据 库号或关键字 查询
序列查询
பைடு நூலகம்
基因结构分析
ProSplicer查询结果
查询NOX1基因:
4.结果表明该ORF编码的蛋白是属于BTB家族。
基因开放阅读框/基因结构分析识别工具
ORF Finder BestORF GENSCAN Gene Finder FGENESH GeneMark GLIMMER /gorf/gorf.html /all.htm /GENSCAN.html /tools/genefinder/ /all.htm /GeneMark/ /genomes/MICROBES/ glimmer_3.cgi /software/glimmer /all.htm NCBI Softberry MIT Zhang lab Softberry GIT Maryland 通用 真核 脊椎、拟南芥、玉米 人、小鼠、拟南芥、酵母 真核 原核 原核
基因结构分析
基因开放阅读框/基因结构分析工具
对基因组序列的读码框区域进行预测
NNSplice /seq_tools/splice.html Web
NetGene2
SPL/SPLM/RNASPL/FSPLICE
http://www.cbs.dtu.dk/services/NetGene2/
可信概率、 得分值
基因预测
GENSCAN输出结果:图形
exon1 exon2 exon3 exon4
exon5
基因预测
ORF识别: GenomeScan
/genomescan.html
提交待分析序列
提交同源蛋白质序列
基因预测
GenomeScan输出结果:文本
/spidey
• NCBI开发的在线预测程序 • 用于mRNA序列同基因组序列比对分析
基因结构分析
Spidey序列提交页面
序列在线提交形式:
界面中有两个窗口:
• 上方窗口用于输入基因组序列(直接粘贴序列或用Genbank ID/AC号) • 下方窗口用于输入cDNA/mRNA序列(直接粘贴序列或用Genbank ID/AC号)
GeneSplicer SplicePredictor
分析mRNA/cDNA的外显子组成
GeneSeqer Spidey Sim4 BLAT BLAST FASTA /cgi-bin/gs.cgi /spidey http://gamay.univ-perp.fr/analyse_seq/sim4 / /~kent/src/unzipped/blat/ ftp:///BLAST/Executables ftp:///pub/fasta/win32_fasta/fasta34t21b5d.zip Web/Linux Web Web/Linux Linux
5’端到3’端 第一位起始: ATG AGT ACC GCT AAA TTA GTT AAA TCA AAA GCG ACC AAT CTG CTT TAT ACC CGC 第二位起始: TGA GTA CCG CTA AAT TAG TTA AAT CAA AAG CGA CCA ATC TGC TTT ATA CCC GC 第三位起始: GAG TAC CGC TAA ATT AGT TAA ATC AAA AGC GAC CAA TCT GCT TTA TAC CCG C
3’端到5’端 第一位起始: GCG GGT ATA AAG CAG ATT GGT CGC TTT TGA TTT AAC TAA TTT AGC GGT ACT CAT 第二位起始: CGG GTA TAA AGC AGA TTG GTC GCT TTT GAT TTA ACT AAT TTA GCG GTA CTC AT 第三位起始: GGG TAT AAA GCA GAT TGG TCG CTT TTG ATT TAA CTA ATT TAG CGG TAC TCA T
如何判断DNA序列的单一基因产物NCBI ORF finder
• 在没有其它信息的前提下,DNA序列可以按 六种框架阅读和翻译(每条链三种,对应 三种不同的起始密码子)。ORF识别包括检 测这六个阅读框架并决定哪一个包含以启 动子和终止子为界限的 DNA序列而其内部不 包含启动子或终止子,符合这些条件的序 列有可能对应一个真正的单一的基因产物。
综合 综合 综合 人 线虫 拟南芥
/tigr-scripts/tgi/splnotes.pl?species=human .tw/ .au/altExtron
/~kent/intronerator/altsplice.html /tdb/e2k1/ath1/altsplicing/splicing_variations.shtml
限 制 酶
目标基因 传统分子生 物学方法 现代生物信 息学方法
重组 基因
BLAST
细胞转化
宿主菌
Gene family Or Protein Family
几分钟的时间
几周的时间 蛋白质分离纯化及性质测定
Function annotation
以Blastx为例: