大规模表达序列标签测定及分析(精)
禾谷镰刀菌基因组学研究进展

禾谷镰刀菌基因组学研究进展张大军,邱德文,蒋伶活*(中国农业科学院植物保护研究所植物病虫害生物学国家重点实验室,北京100081)摘要 禾谷镰刀菌是小麦和大麦生产上一类重大的病原真菌。
禾谷镰刀菌全基因组测序的完成,为禾谷镰刀菌功能基因的发掘提供了十分有利的信息。
简述了禾谷镰刀菌在基因组学,包括比较基因组学和功能基因组学等领域的研究进展。
关键词 禾谷镰刀菌;比较基因组学;功能基因组学中图分类号 S188 文献标识码 A 文章编号 0517-6611(2009)17-07892-03R e se a rch Pro g re s s on th e G e n om ic s o f Fusariu m g r a m inear u m ZHANG D a -ju n e t a l (S ta te K ey L abo ra to ryfo r B io logy o f P lan t D isea se an d In sect P es ts ,In s titu te o f P lan t P ro tection,C h in ese A cade m y o f A gr icu ltu r-a l S cien ces ,B e ijin g 100081)A b s tra c t Fu sarium g r am in earu m is a m a jo r fu n ga l pa th og en on w h ea t an d bar le y produ ction.T h e com p le tion o f F .g ra m inear um gen om ic sequ en cin g prov i de s va lu ab le in form a tion fo r s tu dy in g th e f u n ction a l gen e s o f Fu sariu m g r am i n ear um.T h e recen t re se arch p rog ress on th e g en o m ics o f Fusarium gra m inearu m w e re rev iew ed ,su ch as co m pa ra tive gen om ics an d fu n ction a l gen om ics .K e y w o rd s Fusariu m gra m inear um;C om pa ra tive gen om ics ;F un ction a l g en o m ics基金项目 国家“973”项目(2006CB 101907)。
表达序列标签EST分析及其在林木研究中的应用

林业科学研究 2004,17(6):804~809Forest Research 文章编号:100121498(2004)0620804206表达序列标签(EST)分析及其在林木研究中的应用李 虹1,2,卢孟柱2,蒋湘宁1(11北京林业大学,北京 100083;21中国林业科学研究院林业研究所,北京 100091)摘要:简要叙述了表达序列标签EST技术的原理和流程,综述了EST在研究林木木材形成和其它生物学过程时新基因的发现、基因表达分析和基因芯片方面的应用进展以及在开发林木单核苷酸多态性和简单序列重复等分子标记和构建遗传图谱方面的应用进展,并对其在林木基因组研究中的应用前景进行了展望。
关键词:EST;新基因发现;基因表达;分子标记中图分类号:Q78 文献标识码:A1991年Adams等人从三种人脑组织的cDNA文库中随机挑取609个克隆进行测序,从而得到一组人脑组织的表达序列标签EST(ex pressed sequence tags),并将其与数据库进行序列同源性对比,结果表明:该组EST中有36个代表已知基因,337个代表未知基因,这是关于EST技术应用的首次报道,并首次提出了EST的概念[1]。
随着人类基因组计划的顺利进行,EST技术首先被广泛应用于寻找人类新基因,绘制人类基因组图谱,识别基因组序列编码区等研究领域,之后又被广泛应用于植物基因组研究[2]。
随着EST测序的飞速发展,到2003年6月,美国国家生物技术信息中心(NC BI)的EST数据库中(dbEST)(http:ΠΠw w w.ncbi.nlm.nih.g ovΠdbESTΠindex.html)已录入的来自不同物种的不同组织的EST共有17291123条,其中人和鼠的最多。
EST也被广泛应用于新基因的发现、基因鉴定、基因克隆、构建基因组图谱、基因定位分析、基因表达分析等方面。
在植物方面,除了拟南芥(Arabidopsis thaliana(L.)Heynh.)、水稻(Oryza sativa L.)、小麦(T riticum aesti2 vum L1)、大麦(Hordeum vulgare L.)、大豆(G lycine max(L.)Merr.)、玉米(Zea mays L.)、棉花(G os2 sypium herbaceum L1)等模式植物和农作物以外,近年来也开展了一些木本植物的EST研究,首先报道的是火炬松(Pinus taeda L.)EST分析,随后是杂交杨(Populus tremula L.×P.tremuloides M ichx.)和毛果杨(P.trichocarpa‘T rich obel.’)等其它林木。
大规模基因组测序中的信息分析-文档资料26页

Neural Network Procedure
x
l-1
y
•W =(l)synaptic weight vector of a neuron in layer l
(l)
• =threshold of a neuron in layer l •V =(vl)ector of net internal activity levels of neurons
exon
intron
exon
-------------G U -----------------AG -------------
pseudosplicing sites ---------------GU ------------------AG --------------
Numbe of splicing sites and pseudosplicing sites of testing group
Statistical method and Sequence Alignment Method
eneven positional base frequence (D value)
Neural network
• predicting the splicing sites
Fractal dimension of exons and introns Complexity analysis
• How many different patterns are there in the area of the different DNA sequence?
Method and Techniques in Cryptology
--- Coincident Indexs --- Unicity Distance
表达序列标签(EST)分析及藻类EST文库的研究进展

表达序列标签(EST)分析及藻类EST文库的研究进展陈卓;刘杰;钱万强;孙杰;沈继红;林学政;王能飞【期刊名称】《安徽农业科学》【年(卷),期】2009(037)005【摘要】随着生物信息学的发展,表达序列标签(EST)在基因组作图、克隆基因、新基因的识别、蛋白质组研究等方面具有重要作用.笔者简要介绍了EST技术的原理及其在构建遗传图谱、分离与鉴定新基因等方面的应用.综述了藻类FAST文库的研究现状,并时对EST的应用前景进行了展望.【总页数】4页(P1934-1936,1971)【作者】陈卓;刘杰;钱万强;孙杰;沈继红;林学政;王能飞【作者单位】青岛科技大学生物工程与技术系,山东青岛,266042;青岛科技大学生物工程与技术系,山东青岛,266042;科技部基础研究管理中心,北京,100662;青岛农业大学理学与信息学院,山东青岛,266109;国家海洋局第一海洋研究所生态中心,国家海洋局海洋生物活性物质重点实验室,山东青岛,266061;国家海洋局第一海洋研究所生态中心,国家海洋局海洋生物活性物质重点实验室,山东青岛,266061;国家海洋局第一海洋研究所生态中心,国家海洋局海洋生物活性物质重点实验室,山东青岛,266061【正文语种】中文【中图分类】S188【相关文献】1.大豆表达序列标签(ESTs)研究进展 [J], 崔佳欣;孟军;朱荣胜2.基于表达序列标签(EST)的基因克隆和基因表达分析研究进展 [J], 杨克强;王跃进;张今今;王西平;张剑侠;万怡震3.西洋参cDNA文库构建及表达序列标签(EST)分析 [J], 陈士林;孙永巧;宋经元;李滢;李晨吉;胡松年;李西文;姚辉;张晓伟4.新疆雪莲全长cDNA文库构建及表达序列标签(ESTs)特性分析 [J], 王博;艾秀莲;王志方;李芳;罗明5.小麦条锈菌cDNA文库构建和表达序列标签(ESTs)分析(英文) [J], 张永红;屈志鹏;郑文明;王艳飞;徐亮胜;赵杰;黄丽丽;康振生因版权原因,仅展示原文概要,查看原文内容请购买。
大规模平行测序技术_MPSS_研究进展

新技术新方法大规模平行测序技术(MPSS)研究进展陈杰*(第三军医大学新桥医院肿瘤科,重庆400037)摘要大规模平行测序技术(massively par allel signatur e sequencing,MPSS)是以DNA测序为基础的大规模高通量基因分析新技术,通过标签库的建立、微珠与标签的连接、酶切连接反应和生物信息分析等步骤,获得基因表达序列.MP SS具有能测定表达水平较低、差异较小的基因,不必预先知道基因的序列,自动化和高通量等特点,是值得推广的技术.关键词基因,表达分析,大规模平行测序学科分类号Q34311,Q78大规模平行测序技术(massively parallel signature sequencing,MPSS)是Brenner等[1]于2000年建立,由美国Lynex公司(www.lynex. com)将其商品化的一种基因克隆新技术.在发明之初就与TAKARA公司联手向全球推广.其核心技术分别由MegaClone、MPSS和生物信息分析三部分组成.是基于序列分析技术的高通量、高特异性和高敏感性的基因分析技术.本文就最新建立的大规模平行测序技术做简要介绍,并比较该技术与其他几种常用技术的优缺点.1MPSS概况基因药物及相关诊断试剂开发的首要条件是要找出导致疾病或与疾病相关的基因.人类基因组共有1万至5万个基因,而在人体的任一器官、任一时刻,大约有1万至115万个基因在同时起作用.找出与疾病直接相关的基因是一个复杂的过程,涉及到基因表达水平的测定、蛋白质表达、抗体产生和测试,以及复杂的实验设计和数理统计.而其中最重要的一个环节是准确有效地检测基因在不同样品中表达水平的差异.MPSS是以基因测序为基础的新技术,其方法学基础是一个标签序列(10~20bp)含有能够特异识别转录子的信息,标签序列与长的连续分子连接在一起,便于克隆和序列分析.通过定量测定可以提供相应转录子的表达水平,也就是将mRNA 的一端测出一个包含10至20个碱基的标签序列,每一标签序列在样品中的频率(拷贝数)就代表了与该标签序列相应的基因表达水平,所测定的基因表达水平是以计算mRNA拷贝数为基础,是一个数字表达系统,只要将病理和对照样品分别进行测定,即可进行严格的统计检验,能测定表达水平较低、差异较小的基因,而且不必预先知道基因的序列,该技术的特点是基因表达水平分析的自动化和高通量.大规模平行测序技术,其基本方法是从生物样品中提取mRNA,将mRNA分子转换成cDNA,通过固相克隆将该cDNA均匀地加载到特制的小分子载体表面,然后在小分子载体上进行大量的PCR扩增.将所有cDNA游离的一端进行精确测序产生16至20个碱基.每一特定序列在整个生物样品中所占的比例,就代表了含有该cDNA基因在样品中的相对表达水平.该技术能将一个生物样品中几乎所有表达了的基因全部分别克隆到特制的小分子载体上,然后把几十或上百万个小分子载体放进一个特殊的反应系统内,使所有小分子载体都排列在一个平面上,然后将带特殊荧光标记的G、A、T、C单核苷酸按顺序分别加入反应体系中,分别与小分子载体上的cDNA进行分子杂交,每次分子杂交后将所有小分子载体进行激光扫描照相.当加入G时,有特殊荧光的小分子载体上所载的*通讯联系人.Tel:023*********,E2mail:jazz0331@收稿日期:2004202210,接受日期:2004203228cDNA在这个碱基位置上就是G,当加入A时有荧光,则这个位置就是A,以此类推,只需经过4次反应4次激光扫个碱描照相就可将上百万个cDNA 同时将这一位置的序列测出[2].该技术的特点是:a1不必事先知道基因的序列,适用于任何生物体及任何性状;b1基因组覆盖面高,能测量出样品中几乎所有表达了的基因; c1基因表达水平的测量是通过直接计算样品中cDNA的拷贝数目,属于非连续变量,所以只要有病理和正常个体(或组织)两个样品即可以进行严格的统计检验,能有效地检测差异性中等或较小的基因;d1实验效率高,只要两个星期即可获得几十万个克隆的16至20个碱基序列.该技术的关键是验证数据问题,即如何确定转录子和基因表达水平与标签序列产生的数据之间的关系.对不同的基因使用正确的标签序列,如果基因与标签序列之间是非特异性和不明确的都将会产生分析错误.2MPSS基本步骤首先用生物素标记的寡核苷酸引物(biotin2 labelled oligo2dT primer)将来自细胞或组织的mRNA合成为cDNA双链(图1)[3].Dp nÒ限制性内切酶(酶切位点为GATC)消化cDNA片段,利用标记的生物素纯化消化的cDNA片段.将纯化的cDNA片段克隆入包含有32bp序列的标签(tag)载体中,并通过标签上的PCR引物扩增插入片段.酶切消化线性化PCR产物,生成含cDNA片段与32bp标签相连接的产物.将cDNA模板连接到直径为5L m的微球体上./克隆0的方法是利用人工设计长度不同的两类互补寡核苷酸(tag和anti2tag),分别将cDNA与tag连接,anti2tag和微球体连接之后,再将cDNA模板通过tag和anti2tag杂交连接与微球体连接起来.为了能装载下细胞内所有的cDNA模板(若以310 ~410个基因计算),寡核苷酸的数量至少应该要比模板的量多100倍以上,为此Brenner等设计了1167@107个长32bp的寡核苷酸片段,这样可以保证生物体所有不同的cDNA模板都能与不同的寡核苷酸相连接,而且每一个微球体上也可承载104 ~105个相同的cDNA拷贝[3].与32bp标签序列互补的序列(anti2tag)杂交连接,而anti2tag预先已经通过共价键与直径为5L m的微球体连接,这样含cDNA片段与32bp 标签相连接的序列就与微球体相结合.cDNA序列测定,通过连接接头和ÒS型限制性酶BbvÑ,进一步消化结合在微球体上cDNA模板,BbvÑ能在距识别位点9个碱基和13个碱基的位置切割cDNA双链,并在cDNA模板上产生4个碱基末端.Fig11Atta chment of tags to cDNAs[3]图1cDNA片段与标签及微球体的结合[3]洗脱除去寡核苷酸接头,经过BbvÑ酶切后的cDNA模板,进入下一轮分析.分析所得到的17张荧光显微照片,就可以读出微球体阵列中每一个微球体上长度为17bp的cDNA模板序列(图2)[3].Fig12Deter mine the17mer2signatur e of ea ch cDNA2bead[3]图2每个cD NA2bead序列中17bp标签的产生[3]3MPSS应用根据MPSS技术的原理可以知道,MPSS一方面可提供某一cDNA在体内特定发育阶段的拷贝数,另一方面还可测定出相应cDNA17bp的序列,所以这就为在转录水平上进行基因表达分析提供了强有力的定性和定量手段,很明显,这一技术首先可以应用于不同丰度基因的差异表达分析,制作基因转录图谱,这无疑将加速新基因克隆和基因功能的分析.MPSS所获得的基因序列可提供PCR 引物,可通过比较GenBank EST数据库等进行基因定位,也可转化为分子标记构建遗传图谱等等,因此该技术可广泛用于动植物体分类学和遗传学,功能基因组学,蛋白质组学等研究.Hoth等[4]用MPSS克隆出细胞分裂素上调基因823和下调基因917.Christensen等[5]用MPSS 分析了单叶ROB基因家族的保守亚群和发育调节基因.Jongeneel等[2]用MPSS分析了HB4a(正常乳腺上皮细胞)和H CT2116(结肠腺癌细胞)两株细胞的转录子特征.每株细胞获得了107个序列标签,建立了一个基因表达短标签的分析平台.每个细胞株单拷贝表达基因数量为10000~15000之间.两株细胞中绝大多数转录子都可以在已知基因和多聚A变异体上找到对应的位置,从表达序列标签上克隆的基因,大约8000个两株细胞能公共表达,而6000个分别特异表达.Potschka等[6]以大鼠颞叶癫痫模型为研究对象,采用MPSS技术克隆大鼠癫痫特异表达基因,结果提示,在海马回中有263个特异表达基因,其中,最有意义的是知觉早期基因Homer1A,其功能与谷氨酸受体修饰有关,在癫痫大鼠海马回中过表达.Hoth等[7]在研究基因组ABA(phytohormone abscisic acid,ABA)反应基因在拟南芥(Ara bidopsis thalia na)和abi121突变株中的表达差异时,应用了MPSS技术,结果提示,在ABA处理的野生株中发现的1 354个上调和下调基因,在这些ABA反应基因中大多数编码信号传导组分.在abi121突变的对ABA无反应的对照组中,8415%的克隆基因表达减弱,619%基因表达消失,而816%的基因仍然有一定的调节作用.因此作者认为与其他几种基因表达分析方法相比较,MPSS具有高度特异性和敏感性,是在拟南芥野生株中克隆到大量ABA反应基因的主要手段.4MPSS技术的特点目前,应用于基因克隆的技术有DNA芯片(DNA microarrays)、基因表达系列分析(serial analysis of gene expression,SAGE)、定量PCR (quantitative RT2PCR)、差异显示RT2PCR (differential2display RT2PCR)、抑制消减杂交(subtractive suppress hybridization,SSH)和大规模平行测序(massively parallel signature sequencing, MPSS)等方法.每一种方法,都有自身的优点和不足.考察每一种方法的优劣,应该从该技术的特异性、敏感性、可信性、技术难度和运作成本等方面考虑.尤其是建立的基因表达数据库是否有利于下一步生物信息学的分析.基于序列分析的技术有基因表达系列分析、大规模平行测序(MPSS)和表达标签序列分析等方法.相对而言,基于杂交技术的DDRT2PCR、SSH 和RNA点杂交等技术具有可靠、前期操作简单、通量低、后期生物信息学处理较容易、实验成本低等特点.而基于序列分析的SAGE、EST和MPSS 等具有自动化程度高、通量大、生物信息学处理困难和运作成本高等特点.所以在整体基因和基因组分析中很难说那一种技术占有绝对的优势,研究者可以根据各自的实验目标选择一种适当的方法.MPSS分析系统对基因表达分析过程,诸如微球体阵列的制作,反应液的供排、各种反应条件的控制,图像的处理和数据的分析已经完全自动化,能够在很小的一块微球体阵列上,通过常规的分子生物学手段:连接、酶切、萤光成像等简单几个步骤就可以同时分析数以万计的基因数目,这大大超过了基因的EST、RNAse保护分析、DDRT RT2 PCR分析(这几种方法一次只能检测很少的基因表达情况),甚至超过了SAGE的一次性分析能力,同时不需要耗费时间做大量的PCR实验,不需要对cDNA模板做特殊的处理,也不用对探针序列进行提前选择,因此MPSS技术分析样品基因表达的操作简便,速度快,时间短.更为重要的是MPSS可根据荧光信号对基因表达水平做定量的分析,能提供基因末端序列信息,这是MPSS与RT2 PCR、SAGE等常规方法不同之处.另外,MPSS 对基因末端序列与常规测序不同的是,它不需要进行基因片段的分离、克隆再逐一测序,而是具备了cDNA芯片、cDNA微阵列荧光分析法直接读出序列的优点,可同时获得大量cDNA末端序列,从而简化了测序过程,这符合后基因组时代基因功能分析的高通量、自动化、微型化的要求[8,9].MPSS与基因芯片技术相比较,有下列优点: a1可以避免在cDNA芯片技术中出现的高度同源序列的交叉杂交.因此可以保证基因的高度特异性.9712%的标签中,17bp长度的标签已经足够鉴别基因组中相关的基因.如此高的鉴别率, cDNA芯片技术很难达到;b1MPSS的高分辨率可以检测很低表达水平的基因;c1MPSS技术检测基因不需要预先知道该基因的相关信息,可以应用于任何生物体的基因表达检测,而cDNA芯片技术需要将已知基因片段作为探针固定在片基上[10].当然该技术同DNA芯片技术一样,需要较为昂贵的硬件和相配套的软件协同运做.目前国内外的相关应用报道较少,因此目前还亟需降低仪器检测的成本,加强推广和普及工作.总之,MPSS技术是基因表达定性和定量研究的一种有效工具,它能在短时间内检测细胞或组织内全部基因的表达情况,并能通过与已知基因数据库进行比对,定量显示出基因在细胞或组织内的表达状况,是功能基因组研究和基因发现的有力工具,对于致病基因的识别、药物在组织中的药效分析、揭示基因与疾病之间的传导通路.揭示基因在疾病中的作用都是非常有价值的,而这些与疾病相关的基因将是非常有价值的药靶.随着MPSS技术的不断发展,相信该技术必将在各种生物基因组功能方面及其相关领域研究中发挥巨大的作用.参考文献1Brenner S,Johnson M,Bridgham J,et al.Gene expression analysis by massively parallel signature sequencing(MPSS)on microbead arrays.Nat B iotechnol,2000,18(6):630~6342Jongeneel C V,Iseli C,Stevenson B J,et prehensive sampling of gen e expression in human cell li nes with massivelyparallel signature sequencing.Proc Natl Acad Sci USA,2003,100(8):4702~47053J eannette R,Eddy B,Jingzhong L,et al.Massively parallel signature sequencing(MPSS)as a tool for in2depth quanti tative gene ex pres sion profiling in al l organisms.Briefings i n Functional Genomics and Protemics,2002,1(1):95~1044Hoth S,Ikeda Y,Morgante M,et al.Moni toring genome2wide changes in gen e expression in response to endogenous cytokinin reveals targets i n Arabidop s is thaliana.FEBS Lett,2003,554(3):373~3805Christensen T M,Vej lupkova Z,Sharma Y K,et al.Conserved subgroups and developmental regulation in the monocot rop gene family.Plant Physiol,2003,133(4):1791~18086Potschka H,Krupp E,Ebert U,et al.Kindling2inducedoverexpressi on of hom er1A and i ts functional i mplications for epileptogenesi s.Eur J Neurosci,2002,16(11):2157~21657Hoth S,Morgante M,Sanchez J P,et al.Genom e2wide gene expression profiling in Arabidop s is thaliana reveals new targets of abscisic acid and largely impaired gene regulation in the abi121 mutant.J Cell Sci,2002,115(24):4891~49008Pollock J D.Gene expression profili ng:methodological challenges, results,and prospects for addiction research.Chem Phys Lipids, 2002,121(1~2):241~569Brenner S,Williams S R,Vermaas E H,et al.In vitro cloni ng of complex mixtures of DNA on microbeads:physical separation of differentially ex pres sed cDNAs.Proc Natl Acad Sci USA,2000, 97(4):1665~167010Blohm D H,Guiseppi2Elie A.New developments in microarray technology.Curr Opin B iotechnol,2001,12(1):41~47A Novel Gene Identification Approach:Massively ParallelSignature SequencingCHEN Jie*(C a ncer T reatmen t Center,Xin qiao Hos p ital,The Third Military Medical University,Chongqing400037,China)Abstr act Massively parallel signature sequencing,MPSS,is an open platform that reveals the expression level of virtually all genes expressed in a sample by counting the number of individual mRNA molecules produced from each gene.The MPSS process involves cloning each mRNA molecule onto the surface of a5L m bead.T he DNA combitag sequence is attached to a fragment of cDNA.The cDNA library is hybridized to beads.After hybridization,each of the beads displays amplified copies of one and only one starting mRNA molecule.MPSS has a routine sensitivity of a few molecules of mRNA per cell and the results are in a digital format that simplifies data management and analysis.MPSS results will be particularly useful for generating the type of complete data sets that will help to identify the functionally important genes in the sample of interest.Key words gene,expression analysis,massively parallel signature sequencing(MPSS)*Corresponding author.T el:86223268755646,E2mail:jazz0331@Received:February10,2004Accepted:March28,2004。
转录组学研究方法

In Situ Oligo Synthesis
Ø Photosynthesis
Ø Ø
原位合成芯片
Ø Oligo Arrays
Planer surface Microfluidics chip
Ø E-field synthesis
Microfluidics
微流体芯片 Ø Plastics Ø Ceramics Ø Silicon Ø Other materials
● Digital Gene Expression Displayer (DGED)
● cDNA xProfil无理想匹配
InterproScan
较好匹配
域的注释
析
无理想匹配
New sequences
基因功能分类
◆
手工分类
大部分以Adams 95年的文章中的采用分类体系为标准。
【Adams. MD, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995 377(6547 Suppl):3-174 】
RT-PCR
RT-PCR是将RNA的反 转录(RT)和cDNA的 聚合酶链式扩增(PCR )相结合的技术。首 先经反转录酶的作用 从RNA合成 cDNA,再 以cDNA为模板,扩增 合成目的片段。
3’ RACE
• 以mRNA的 polyA为锚定
5’ RACE
• 原理上比3’RACE要 稍微复杂 • 要点: 逆转录酶 MMLV合成cDNA具 有加尾特性,即在 合成的cDNA链3’加 上3-4个dCTP,而且 当存在帽子结构时 该酶的加尾活性最 高 • 然后以这段polyC为 锚定
基因表达标签测序技术
基因表达标签测序技术基因表达标签测序(Tag profiling)技术是基于Illumina高通量测序平台的全基因组表达谱研究技术,其原理是使用每个转录本3’端一段特定的21bp标签序列来表征相应转录本的表达水平,利用Illumina高通量测序技术获得测序文库中所有标签的序列信息,进一步通过生物信息学分析比对,鉴定这些标签序列所代表的基因,以及根据相同标签序列出现的频率计算该基因的表达水平,同时还能够比较不同样品间这些基因表达水平存在的差异。
相对于传统的基于杂交技术的基因芯片分析平台,基因表达标签测序能够提供更加精确的数字化信号,更高的基因表达数据检测通量以及更广的检测范围。
同时,由于不需要预先针对已知序列设计探针,它能够直接用于任何物种的全基因组表达谱分析,在检测未知转录本,稀有转录本以及反义转录本等方面具有无可比拟的优势。
技术优势¾数字化信号:基于高通量测序技术的表达谱分析,直接测定每个转录本标签序列,通过标签序列计数来确定基因表达量,极大地提高了定量分析的精确度。
不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题,因此样品间细微的表达差异也能够被检测出来。
¾高灵敏度:一般认为,单个细胞中的转录本数量是35万个,而一个通道测序至少产生400万有效标签数,这意味着即使是单拷贝的稀有转录本,在一次实验中也能平均被测到12次,非常利于检测低丰度转录本。
¾任意物种的全基因组分析:不需要预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行全基因组表达谱分析,同时能够检测未知基因,发现新的转录本。
所得到的数据能够使用现有的基因组数据库注释,当现有数据库更新后,只需对原始数据进行重新注释即可进行新的分析。
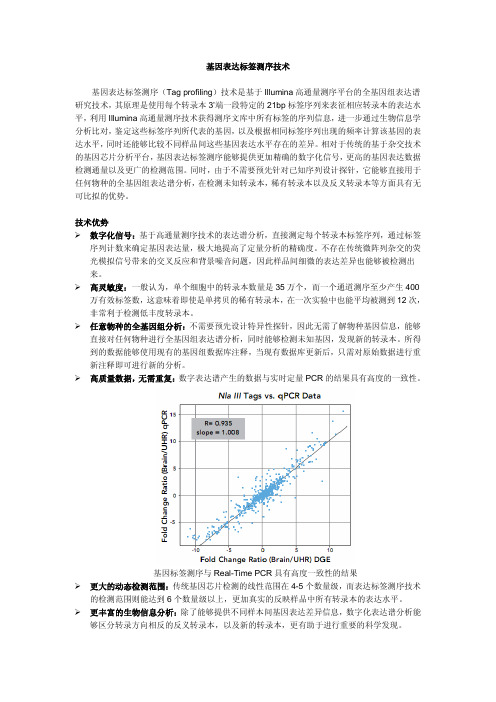
¾高质量数据,无需重复:数字表达谱产生的数据与实时定量PCR的结果具有高度的一致性。
基因标签测序与Real-Time PCR具有高度一致性的结果¾更大的动态检测范围:传统基因芯片检测的线性范围在4-5个数量级,而表达标签测序技术的检测范围则能达到6个数量级以上,更加真实的反映样品中所有转录本的表达水平。
10第十一章DNA序列测定和分析

A > C 的反应
还有一种反应(A>C),必要时可用来参考。在 90℃下,用1.2mol/L NaOH处理,可使A位点发生剧烈的 断裂反应,而C位点的断裂反应较微弱。
第19页,共81页。
四、测序策略
鸟枪法
鸟枪法又称随机法,它是先将大片段DNA经酶切或 超 声 处 理 , 切 割 成 适 合 测 序 大 小 的 片 段 ( 如 500 ~ 800bp),然后亚克隆到M13载体上,得到单链DNA后测 序,再根据重叠的部分推测出整个大片段的序列。
速、大规模测序提供了新的思路和方法。
第24页,共81页。
1. Roche(454)GS FLX sequencer
Roche公司收购454公司的测序仪并经改造升级, 该测序仪最早的商业化产品于2004年推出。454测P载样。
(4)测序。
第12页,共81页。
自动化测序的输出信号
第13页,共81页。
三、Maxam-Gilbert 化学直读法
这是一种基于DNA降解的方法。此法的原理是首 先 将 待 测 序 的 DNA 片 段 一 端 用 放 射 性 标 记 , 然 后 使 DNA链在特定碱基处断裂。控制条件使每个DNA分子 只发生一处断裂,就可以得到一系列只差一个核苷酸 的DNA片段,跑电泳后可得出序列。
第十一章 DNA序列测定和分析
一、加减法
二、 Sanger 双脱氧链终止法 三、 Maxam-Gilbert 化学直读法
四、测序策略
五、第二代测序技术
六、杂交测序与DNA芯片技术
七、表达序列标签
八、 DNA 序列分析
第1页,共81页。
一、加减法测序
以 待 测 DNA 为 模 板 , 加 一 同 位 素 标 记 的 短 链 引 物 , 在 4×dNTP存在下,用DNA聚合酶Ⅰ催化合成各种随机长度的 产物。将模板及合成的产物分为“加法组”和“减法组”, 加法组和减法组又各分为4组。“加法组”中的每一小组只加 一种dNTP,4组各加不同的dNTP。以加dATP组为例,当前 面合成的随机长度的DNA下一个核苷酸该加A时,可以将A加 上;如果正好以A结尾,而下一个不该加A时,则链保持不变; 如果不以A结尾,下一个又不该加A时,则利用DNA聚合酶Ⅰ 的3’→5’外切活性逐个切除已合成的核苷酸,直到遇到A为止。 最终加dATP组的每一条新合成的链都是以A结尾,整个组中 各个A处结尾的链都有。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
高密度寡核苷酸cDNA 芯片或cDNA微阵列是一种新的大规模检测基因表达的技术,具有高通量 分析的优点。在许多情况下,cDNA芯片的探针来源于3'EST (Duggan et al., 1999),所以EST序列 的分析有助于芯片探针的设计。
Serial analysis of gene expression (SAGE) 技术流程
ESTs与基因预测
由于EST的一个基因的部分序列。使用合适的比 对参数,大于90%的已经注释的基因都能在EST库中检测到(Bailey et al., 1998)。ESTs可以做为其它基因预测算法的补充,因为它们对预测 基因的交替剪切和3‘ 非翻译区很有效。
什么是 ESTs ?
ESTs(Expressed Sequence tags )是从已建好的cDNA库中随机取出一个 克隆,从5’末端或3’末端对插入的cDNA片段进行一轮单向自动测序,所 获得的约60-500bp的一段cDNA序列。
ESTs与基因识别
ESTs已经被广泛的应用于基因识别,因为ESTs的数目比GenBank 中其它的核苷酸序列多,研究人员更容易在EST库中搜寻到新的基因 (Boguski et al., 1994).
● 在同一物种中搜寻基因家族的新成员(paralogs)。
● 在不同物种间搜寻功能相同的基因(orthologs)。 ● 已知基因的不同剪切模式的搜寻。【注:不过很难确定一个新 的erg et al., 1997)】
利用ESTs大规模分析基因表达水平
因 为研究癌症的分子机理,美国国家癌症研究所NCI的癌症基因组解析计划(Cancer Genome Anatomy Project , CGAP)构建了很多正常的s与SNPs
来自不同个体的冗余的ESTs可用于发现基因组中转录区域存在的SNPs。 最近的许多研究都证明对ESTs数据的分析可以发现基因相关的SNPs (Buetow et al., 1999;Garg et al., 1999; Marth et al., 1999; Picoult-Newberg et al., 1999) 。 应注意区别真正的SNPs和由于测序错误( ESTs为单向测序得来,错误率 可达2%)而引起的本身不存在的SNPs。解决这一问题可以通过: ● 提高ESTs分析的准确性。 ● 对所发现的SNPs进行实验验证。
实 验 步 骤 较 长 要 求 较 高
反转录 酶切 连接
测序
单条测序==对30-40条EST测序
CGAP网站提供了多种工具用以分析不同间基因表达的差异, 如:● Digital Gene Expression Displayer (DGED) ● cDNA xProfiler ◆ 基因表达系列分析(Serial Analysis of Gene Expression, SAGE) 基因表达系列分析是一种用于定量,高通量基因表达分析的实验方法(Velculescu et al., 1995)。 SAGE的原理就是分离每个转录本的特定位置的较短的单一的序列标签(约9-14个碱基对),这些短 的序列被连接、克隆和测序,特定的序列标签的出现次数就反应了对应的基因的表达丰度。 ◆ DNA微阵列或基因芯片的研究
ESTs与基因图谱的绘制
EST可以借助于序列标签位点(sequence-tagged sites)用于基因图谱的 构建. STS本身是从人类基因组中随机选择出来的长度在200-300bp左右的 经PCR检测的基因组中唯一的一段序列。来自mRNA的3’非翻译区的ESTs 更适合做为STSs,用于基因图谱的绘制。其优点主要包括: ● 由于没有内含子的存在,因此在cDNA及基因组模板中其PCR产物的 大小相同; ● 与编码区具有很强的保守性不同,3’UTRs序列的保守性较差,因此 很容易将单个基因与编码序列关系非常紧密的相似基因家族成员分开。 (James Sikela等,1991年) GeneMap 96‘ 定位了16,000个基于基因的STS(Schuler et al., 1996); GeneMap 98’ 定位了30,000个基于基因的STS(Deloukas et al., 1998),而 且基因图谱随着STS的定位正在不断的更新中。
dbEST 中数据量的增长
20 18 16 14 12 10 8 6 4 2 0
总EST条目 (million)
199319951源自9719992001
时间(年)
●
93年前ESTs数据收录于GenBank, EBI和DDBJ。
● 1993年NCBI(National Center of Biotechnology Information)建立 了一个专门的EST数据库dbEST来保存和收集所有的EST数据。
大规模表达序列标签(EST)测 定及分析
中山大学生科院
主要内容
• 什么是EST? • EST的应用 • EST序列测定及分析过程 • 实例:家猪脑组织EST分析
ESTs的来源
上世纪80年代,对cDNA序列进行大规模测序的想法就曾提出,但对此 一直存在争论,有人认为这种方法能发现成千上万的新基因;而反对者则认 为cDNA序列缺少重要的基因调控区域的信息。90年代初Graig Venter 提出 了EST的概念,并测定了609条人脑组织的EST,宣布了cDNA大规模测序的时 代的开始 (Adams et al., 1991)。