自旋轨道耦合计算探索过程分析
自旋轨道耦合矩阵元的单元

自旋轨道耦合矩阵元的单元一、引言自旋轨道耦合(spin-orbit coupling, SOC)是一种重要的物理效应,它是导致许多奇妙物理现象的根本原因。
该效应描述了自旋和轨道角动量在量子力学中的相互作用,其中自旋指粒子的内禀属性,轨道角动量指粒子绕原子核运动的角动量。
自旋轨道耦合在原子物理、凝聚态物理、量子信息学等多个领域都发挥着重要的作用。
自旋轨道耦合矩阵元是研究自旋轨道耦合效应的关键参数。
在本文中,我们将探讨自旋轨道耦合矩阵元的单元,着重介绍自旋轨道耦合矩阵元的物理意义和计算方法。
二、自旋轨道耦合矩阵元的物理意义自旋轨道耦合矩阵元是描述自旋和轨道角动量相互作用的参数。
在量子力学中,自旋角动量和轨道角动量不满足对易关系,因此自旋和轨道角动量不能同时精确测量。
自旋轨道耦合矩阵元描述了自旋和轨道角动量的纠缠程度,其大小和符号决定了粒子的自旋和轨道角动量是如何相互作用的。
举例来说,物理学家可以利用自旋轨道耦合矩阵元来描述氢原子中1s态的自旋和轨道耦合。
在没有自旋轨道耦合的情况下,1s态有两种可能的自旋状态:自旋向上和自旋向下。
当自旋轨道耦合存在时,1s态中的自旋和轨道角动量耦合在一起,形成一个新的量子态,称为自旋轨道耦合态。
自旋轨道耦合态存在三种可能的自旋状态:自旋向上、自旋向下和自旋不定。
自旋轨道耦合矩阵元描述了这三种不同自旋态之间的转换概率和相对相位。
三、计算自旋轨道耦合矩阵元的方法自旋轨道耦合矩阵元的计算方法取决于所研究的体系。
以下介绍两种常见的计算方法。
1.量子力学方法量子力学方法是计算自旋轨道耦合矩阵元的基础方法,它涉及到原子结构理论和核结构理论的综合应用。
该方法用到了量子力学的基本原理和表达式,例如薛定谔方程、包络函数、哈密顿量的各种形式等。
通常需要用到高精度的计算机算法和软件来计算自旋轨道耦合矩阵元。
2.实验方法实验方法使用粒子探测器等实验设备对粒子自旋和轨道角动量的相互作用进行直接测量。
量子力学知识:量子力学中的自旋轨道耦合

量子力学知识:量子力学中的自旋轨道耦合自旋轨道耦合是量子力学中非常重要的一个概念,描述了自旋和轨道角动量之间的相互影响。
在经典力学中,自旋和轨道角动量是分离的量,而在量子力学中,它们之间是相互耦合的。
本文将从自旋、轨道角动量入手,探讨自旋轨道耦合的原理及其在量子力学中的应用。
一、自旋与轨道角动量自旋和轨道角动量是两个不同的概念。
轨道角动量是一个物体在围绕某个中心点旋转时所具有的角动量,而自旋是指某个粒子自身所具有的角动量。
虽然这两者名称相似,但它们的物理性质和测量方式都不同。
轨道角动量可以通过位置和动量算符的组合来描述。
假设一个粒子在坐标(x, y, z)处,其中X、Y、Z是三个方向的运动算符,则该粒子的轨道角动量为:L = (xpy - ypx)i + (zpx - xpz)j + (ypz - zpy)k自旋是一种固有的角动量,粒子表现出来具有像自转一样的角动量。
自旋基本上可以由两个不同的贡献来组成:与电子磁矩相关的轨道自旋和与电子内部结构相关的自旋角动量。
自旋可以被描述为自旋算符S的乘积,其中Sx、Sy和Sz是自旋算符的三个分量。
自旋算符是一个特殊的算符,作用于它所描述的粒子时,可以测量出粒子的自旋。
二、自旋轨道耦合的原理自旋和轨道角动量之间最显著的相互影响就是自旋轨道耦合。
通过自旋轨道耦合,电子的自旋和轨道角动量产生相互作用,从而形成新的能级结构和特别的光谱性质。
自旋轨道耦合的原理可以通过考虑磁场的影响来解释。
磁场描绘了电子在运动的过程中具有的电荷加速度,因此会产生相应的电子自旋和轨道线性动量。
这个磁场的大小与电流的大小成正比,因此可以通过外部的磁场来控制它的大小。
在一个强磁场下,电子会被强制沿着一条定义良好的轨道运动,这个轨道和电子的内部构造相关联,从而与自旋相互作用。
当两个轨道之间的磁场强度发生变化时,这种相互作用就会发生。
某些原子中的电子会沿着一个运动轨道运动,而另一些电子则会改变自己的自旋,从而导致新的态出现。
自旋轨道耦合计算

自旋轨道耦合计算自旋轨道耦合计算是一种重要的物理计算方法,它涉及到原子、分子和凝聚态系统中自旋和轨道的相互作用。
以下将分步骤阐述自旋轨道耦合计算的基本原理和应用。
一、自旋和轨道的基本概念自旋是微观粒子的一个内部自由度,它描述了粒子的自旋角动量。
轨道则是宏观物理中一个物体周围的运动轨迹,而在微观物理学中,轨道是描述电子绕原子核的运动轨迹。
自旋和轨道都是量子态的内在属性,它们的相互作用很强,这就是自旋轨道耦合现象的来源。
二、自旋轨道耦合的作用自旋轨道耦合对于原子、分子和凝聚态系统的性质有着重要的影响。
例如,在磁学、光谱学、量子计算和量子信息等领域中,自旋轨道耦合可以使含有奇异自旋态的物质表现出不同寻常的物理性质。
三、计算方法自旋轨道耦合的计算方法可以分为半经典和量子力学两种方法。
半经典方法基于经典电磁场理论,相对简单,主要适用于原子、分子系统。
量子力学方法则更加广泛,可以处理复杂的凝聚态系统。
具体计算可采用量子化学方法和密度泛函理论等。
四、应用领域1. 量子计算:自旋轨道耦合可以用于永久性化学计算中的生成和操作变量的编码。
2. 拓扑绝缘体:包括量子自旋液体、拓扑半金属和拓扑绝缘体等的物理研究。
3. 磁共振成像:自旋轨道耦合可以用于特定的核磁共振成像模型中,例如,结合单磁子、双磁子和三磁子方法,来进行局部的可视化。
结论:自旋轨道耦合计算是量子物理研究领域的重要方法。
它不仅能够帮助人们研究原子、分子和凝聚态系统的性质,而且还能在量子计算、拓扑绝缘体和磁共振成像等领域中发挥重要作用。
我们相信,在未来的研究中,自旋轨道耦合计算会在各个领域取得更加广泛和深入的应用。
kagome晶格的自旋轨道耦合

kagome晶格的自旋轨道耦合Kagome晶格是一种具有特殊排列结构的晶体,其晶格形状类似于日本著名的Kagome篮子。
最近,科学家们对Kagome晶格的自旋轨道耦合进行了深入研究,并发现了一些令人惊奇的性质。
自旋轨道耦合是指电子轨道运动和自旋运动之间的相互作用。
在一般的材料中,自旋轨道耦合并不明显,然而在Kagome晶格中,由于其特殊的结构,自旋轨道耦合效应显著增强。
在Kagome晶格中,晶格结构由三个相互连接的三角形单元组成。
每个晶格点上具有一个电子自旋,而这些自旋之间的相互作用通过晶格的排列方式和化学键形成。
自旋轨道耦合通过引入晶体中的重元素离子来实现。
这些重元素离子具有较高的自旋轨道耦合常数,导致自旋与电子轨道运动之间的相互作用变得更加显著。
在Kagome晶格中引入重元素,使得自旋轨道耦合变得强烈,从而产生了一些独特的性质。
首先,自旋轨道耦合使得Kagome晶格的自旋基态排列方式发生变化。
在一般的自旋系统中,自旋会通过玻尔兹曼分布随机排列。
然而,在Kagome晶格中,自旋轨道耦合强烈影响自旋基态的排列方式,使得自旋在晶体中形成某种特定的有序性。
其次,自旋轨道耦合使得Kagome晶格呈现出一些特殊的物理性质。
例如,在一般的自旋系统中,自旋之间通常不存在长程的自旋相干关系。
然而,在Kagome晶格中,由于自旋轨道耦合的存在,自旋之间可以发生长程的自旋相干,导致一些非常有趣的量子效应的出现。
最后,自旋轨道耦合还可以导致Kagome晶格中出现一些拓扑性质。
拓扑性质是指物体形状或结构的一种特殊性质,与物体的连续变形无关。
在Kagome晶格中,由于自旋轨道耦合的存在,自旋之间的相互作用可以是非局域的,导致晶格中存在一些特殊的拓扑状态,如拓扑绝缘体等。
总之,Kagome晶格的自旋轨道耦合是一个非常有趣和重要的研究课题。
通过对Kagome晶格中自旋轨道耦合的研究,科学家们对于材料的特殊性质有了更深入的理解,这对于开发新型材料和实现新型电子器件具有重要意义。
自旋轨道耦合的推导

课程作业题目: 自旋轨道耦合的推导姓名:学号:班级:2014年11月8号摘要:本文通过计算电子的进动动能得出自旋轨道耦合公式,并对课本中∆E ls=1这个模糊的问题提出看法。
2关键字:自旋-轨道耦合能;托马斯进动;目录1引言 (4)2关于课本推导的讨论 (4)3自旋同轨道相互作用推导 (5)4参考文献 (7)1 引言在量子力学里,一个粒子因为自旋与轨道运动而产生的作用,称为自旋-轨道作用。
最著名的例子是电子能级的位移。
电子移动经过原子核的电场时,会产生电磁作用.电子的自旋与这电磁作用的耦合,形成了自旋-轨道作用。
谱线分裂实验明显地侦测到电子能级的位移,证实了自旋-轨道作用理论的正确性。
另外一个类似的例子是原子核壳层模型(shell model)能级的位移。
本文根据环形电流公式计算有效磁场来推导相互作用的公式。
2 关于课本推导的讨论在原子物理学课本中130-131面对相互作用公式进行了推导。
推导思路是这样。
电子的自旋轨道耦合能一般都根据电磁学理论得出。
如图1设原子磁矩与磁场之间的夹角是θ。
则原子受力矩使转向的方向,使θ减小。
若θ增加dθ,做功力矩作功dA等于势能W的减小,选取θ=π2,W=0则具有磁矩的原子在磁场中具有能量由此得出, 自旋磁矩为s的电子在磁场中所具有的能量但是电子磁矩是由于它具有轨道角动量。
电子磁矩在磁场中受力矩作用不是使磁矩转向磁场方向, 而是使电子的角动量绕磁场方向作拉摩尔进动, 使电子的动能发生变化。
这和磁性物体在磁场中具有势能的机制有根本区别。
另外考虑到参照系问题。
一般选取实验室坐标系, 在这里就是原子核或原子实, 严格来说应是质心坐标系。
但从原子核坐标系来看, 电子处只有静电场而无磁场, 所以无法用上式来计算∆E ls。
为了解决存在问题, 一般认为上式中的是在电子坐标系中所观察到的磁场, 也就是电子感受到由于其轨道运动产生的磁场, 即原子核绕电子运动所产生的磁场。
然后考虑到电子绕原子核旋转, 有一个加速度, 因此电子坐标系相对于原子核坐标系有一个托马斯进动。
自旋轨道耦合计算探索过程分析
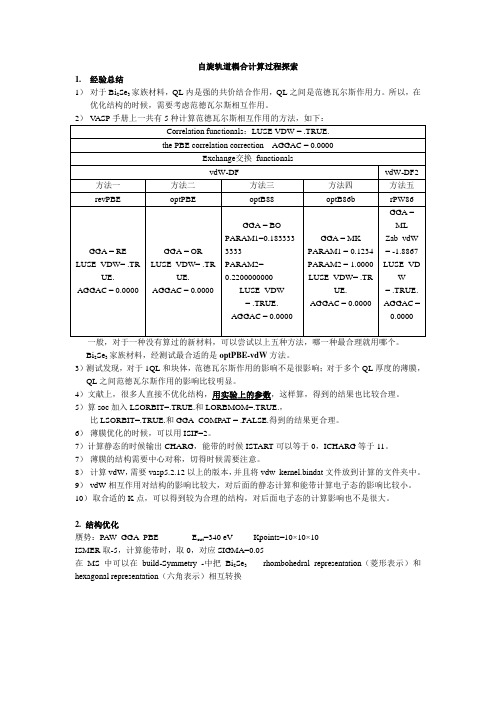
自旋轨道耦合计算过程探索1.经验总结1)对于Bi2Se3家族材料,QL内是强的共价结合作用,QL之间是范德瓦尔斯作用力。
所以,在优化结构的时候,需要考虑范德瓦尔斯相互作用。
一般,对于一种没有算过的新材料,可以尝试以上五种方法,哪一种最合理就用哪个。
Bi2Se3家族材料,经测试最合适的是optPBE-vdW方法。
3)测试发现,对于1QL和块体,范德瓦尔斯作用的影响不是很影响;对于多个QL厚度的薄膜,QL之间范德瓦尔斯作用的影响比较明显。
5)算soc加入LSORBIT=.TRUE.和LORBMOM=.TRUE.,比LSORBIT=.TRUE.和GGA_COMPAT = .FALSE.得到的结果更合理。
6)薄膜优化的时候,可以用ISIF=2。
7)计算静态的时候输出CHARG,能带的时候ISTART可以等于0,ICHARG等于11。
7)薄膜的结构需要中心对称,切得时候需要注意。
8)计算vdW,需要vasp5.2.12以上的版本,并且将vdw_kernel.bindat文件放到计算的文件夹中。
9)vdW相互作用对结构的影响比较大,对后面的静态计算和能带计算电子态的影响比较小。
10)取合适的K点,可以得到较为合理的结构,对后面电子态的计算影响也不是很大。
2. 结构优化赝势:PAW_GGA_PBE E cut=340 eV Kpoints=10×10×10ISMER取-5,计算能带时,取0,对应SIGMA=0.05在MS中可以在build-Symmetry -中把Bi2Se3 rhombohedral representation(菱形表示)和hexagonal representation(六角表示)相互转换图中黑色t 1、t 2、t 3基矢围成菱形原胞,用于计算块体,红色方框包含一个五元层 计算能带的布里渊区高对称点:块体:文献中倒空间高对称点坐标Г(0 0 0)-Z(π π π)-F(π π 0)-Г(0 0 0)-L(π 0 0), 根据正空间和倒空间坐标的转换关系,得到正空间中高对称点的坐标: Г(0 0 0)-Z(0.5 0.5 0.5)-F(0.5 0.5 0)-Г(0 0 0)-L(0 0 -0.5) KPOINTS 20 Line-mode Rec0.0 0.0 0.0 !Г 0.5 0.5 0.5 ! Z 0.5 0.5 0.5 ! Z 0.5 0.5 0.0 ! F 0.5 0.5 0.0 ! F 0.0 0.0 0.0 !Г 0.0 0.0 0.0 !Г 0.0 0.0 -0.5 ! L[通过比较结构,发现Ecut=580,KPOINTS=151515,得到的结构比较靠谱]3. 块体soc 的计算 文献能带结构图:块体(Bi 2Se 3-VASP-GGA-PAW-PBE )我们的结果(未考虑vdW+静态和能带都加soc计算结果与文献基本符合):4.薄膜的计算薄膜:Kpoints=10×10×1计算能带的K点和石墨烯(六角晶胞的)的K点一样:KPOINTS20Lone-modeRec0.66666667 0.33333333 0.0 !K0.0 0.0 0.0 !Г0.0 0.0 0.0 !Г0.5 0.0 0.0 !M考虑薄膜的对称性由MS六角结构,沿(001)方向切割,可以得到两种以Se原子作为表面原子的薄膜,如下图,分别为1QL和3QL的两种切法,右图比左图对称性要更好一些,这一区别在计算过程中会导致巨大的区别,我们通过比较,发现,只有右图的结果,才可以得到合理的结果,尤其是在多个QL的情况。
【方向】VASP自旋轨道耦合计算

【关键字】方向VASP 自旋轨道耦合计算已有4532 次阅读2011-9-13 20:37|个人分类:VASP|系统分类:科研笔记将VASP 的makefile 文件中的 CPP 选项中的 -DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf 这4个选项去掉重新编译VASP才能计算自旋轨道耦合效应。
以下是从VASP在线说明书整理出来的非线性磁矩和自旋轨道耦合的计算说明。
非线性磁矩计算:1)计算非磁性基态产生WAVECAR和CHGCAR文件。
2)然后INCAR中加上ISPIN=2ICHARG=1 或 11 !读取WAVECAR和CHGCAR文件LNONCOLLINEAR=.TRUE.MAGMOM=注意:①对于非线性磁矩计算,要在x, y 和 z方向分别加上磁矩,如MAGMOM = 1 0 0 0 1 0 !表示第一个原子在x方向,第二个原子的y方向有磁矩②在任何时候,指定MAGMOM值的前提是ICHARG=2(没有WAVECAR和CHGCAR文件)或者ICHARG=1 或11(有WAVECAR和CHGCAR文件),但是前一步的计算是非磁性的(ISPIN=1)。
磁各向异性能(自旋轨道耦合)计算:注意: LSORBIT=.TRUE. 会自动打开LNONCOLLINEAR= .TRUE.选项,且自旋轨道计算只适用于PAW赝势,不适于超软赝势。
自旋轨道耦合效应就意味着能量对磁矩的方向存在依赖,即存在磁各向异性能(MAE),所以要定义初始磁矩的方向。
如下:LSORBIT = .TRUE.SAXIS = s_x s_y s_z (quantisation axis for spin)默认值: SAXIS=(0+,0,1),即x方向有正的无限小的磁矩,Z方向有磁矩。
要使初始的磁矩方向平行于选定方向,有以下两种方法:MAGMOM = x y z ! local magnetic moment in x,y,zSAXIS = 0 0 1 ! quantisation axis parallel to zorMAGMOM = 0 0 total_magnetic_moment ! local magnetic moment parallel to SAXIS (注意每个原子分别指定)SAXIS = x y z !quantisation axis parallel to vector (x,y,z),如 0 0 1两种方法原则上应该是等价的,但是实际上第二种方法更精确。
arpes研究自旋轨道耦合

arpes研究自旋轨道耦合
ARPES (Angle-Resolved Photoemission Spectroscopy) 是一种研
究材料电子结构的实验技术,它通过照射样品表面的光子束,观察光电子的发射角度和能量来获取材料的能带结构信息。
自旋轨道耦合是一种在材料中存在的相互作用,它描述了自旋和轨道运动之间的相互影响。
在自旋轨道耦合的材料中,电子的自旋和轨道角动量耦合在一起,产生新的能带结构和自旋态。
利用ARPES技术,可以直接观察到材料中的自旋轨道耦合效应。
通过测量光电子的自旋极化信息,可以确定自旋轨道耦合对能带结构的影响。
同时,由于ARPES技术具有高角分辨率
和能量分辨率,可以准确地测量自旋轨道耦合导致的细微能带结构的变化。
自旋轨道耦合在许多领域都具有重要的应用,例如拓扑绝缘体和自旋电子学。
通过ARPES研究自旋轨道耦合,可以为这些
应用提供重要的材料特性参数,以及对自旋态的深入理解。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
自旋轨道耦合计算过程探索1.经验总结1)对于Bi2Se3家族材料,QL内是强的共价结合作用,QL之间是范德瓦尔斯作用力。
所以,在优化结构的时候,需要考虑范德瓦尔斯相互作用。
一般,对于一种没有算过的新材料,可以尝试以上五种方法,哪一种最合理就用哪个。
Bi2Se3家族材料,经测试最合适的是optPBE-vdW 方法。
3)测试发现,对于1QL和块体,范德瓦尔斯作用的影响不是很影响;对于多个QL厚度的薄膜, QL之间范德瓦尔斯作用的影响比较明显。
4)文献上,很多人直接不优化结构,用实验上的参数,这样算,得到的结果也比较合理。
5)算soc 力廿入LSORBIT=.TRUE.和口LORBMOM=.TRUE.,比LSORBIT=.TRUE.和GGA_COMPAT = .FALSE.得到的结果更合理。
6)薄膜优化的时候,可以用ISIF=2。
7)计算静态的时候输岀CHARG,能带的时候ISTART可以等于0, ICHARG 等于11。
7)薄膜的结构需要中心对称,切得时候需要注意。
8)计算vdW,需要vasp5.2.12以上的版本,并且将vdw_kernel.bindat文件放到计算的文件夹中。
9)vdW相互作用对结构的影响比较大,对后面的静态计算和能带计算电子态的影响比较小。
10)取合适的K点,可以得到较为合理的结构,对后面电子态的计算影响也不是很大。
2.结构优化赝势:PAW_GGA_PBE E cut=340 eV Kpoints=10 W X10ISMER取-5,计算能带时,取0,对应SIGMA=0.05在MS 中可以在build-Symmetry -中把Bi 2Se s rhombohedral representation (菱形表示)和hexagonal representation (六角表示)相互转换图中黑色t i、t2、t3基矢围成菱形原胞,用于计算块体,红色方框包含一个五元层计算能带的布里渊区高对称点:块体:文献中倒空间高对称点坐标r (0 0 O》Z(n n-nn n硼(0 0 0》L( n 0 0)根据正空间和倒空间坐标的转换关系,得到正空间中高对称点的坐标:r (0 0 0-Z(0.5 0.5 0.5)-F(0.5 0.5 0)- r (0 0 0)L(0 0 -0.5)KPOINTS20Lin e-modeRec0.0 0.0 0.0 ! r0.5 0.5 0.5 ! Z0.5 0.5 0.5 ! Z0.5 0.5 0.0 ! F0.5 0.5 0.0 ! F0.0 0.0 0.0 ! r! r0.0 0.0 0.00.0 0.0 - -0.5 ! L[通过比较结构, 发现Ecut=580 , KPOINTS=151515,得到的结构比较靠谱]3. 块体soc的计算文献能带结构图:块体(Bi 2Se3-VASP-GGA-PAW-PBE )0.666666670.333333330.0!K0.0 0.0 0.0 ! r0.0 0.0 0.0 ! r0.5 0.0 0.0 !M考虑薄膜的对称性由MS六角结构,沿(001 )方向切割,可以得到两种以Se原子作为表面原子的薄膜,如下图,分别为1QL和3QL的两种切法,右图比左图对称性要更好一些,这一区别在计算过程中会导致巨大的区别,我们通过比较,发现,只有右图的结果,才可以得到合理的结果,尤其是在多个QL的情况。
>出2 10a我们的结果(未考虑vdW+静态和能带都加soc计算结果与文献基本符合)4.薄膜的计算薄膜:Kpoints=1O X10X1计算能带的K点和石墨烯(六角晶胞的)的K点一样:KPOINTS20Lon e-modeRec用左边结构得到的结果(Bi 2Se3):-0.5\ /^30.0 0.5用右边结构得到的结果(Bi2Se3):1QL —根据块体的数据得到薄膜,分以下两种情况计算:1.不优化结构,scf不加soc, bands加soc2.用块体的参数,加vdW 优化结构,scf不加soc,ba nds加soc1QL —在静态中也加入soc1.不优化结构,scf 禾口bands 力廿入L ORBMOM=.TRUE. ,LSORBIT=.TRUE.2.不优化结构,scf 和bands 加入LORBMOM=.TRUE. ,GGA_COMPA T=.FALSE.3.优化结构,scf 和口bands 力廿入L ORBMOM=.TRUE. ,LSORBIT=.TRUE.4.优化结构,scf 和ba nds 加入LORBMOM=.TRUE. ,GGA_COMPA T=.FALSE.文献结果:上图是没有进行离子弛豫的1QL ~6QL 的Bi2Se3薄膜能带结构5.调试过程错误总结错误 1: VERY BAD NEWS! Internal error in subroutine IBZKPT: Reciprocal lattice and k-latticebelong todifferent class of lattices. Often results are still useful... 48 In ter nal 内部 subrout ine 子程序 Reciprocal 倒数的非常严重的错误! 子程序IBZKPT 中内部错误:倒格子和k 点网格属于不同类型的格子。
通常结果还是有用的。
解决方案: 根据所用集群,修改INCAR 中NPAR 。
将NPAR=4变成NPAR=1,已解决!<a)上图采用optPBE-vdW 泛函进行离子弛豫r r r1QL~6QL 的Bi2Se3薄膜能带结构上图是实验观测的1QL ~6QL ( 12356)的 Bi2Se3薄膜能带结构.MU氢吉点,0.5错误2:in ternal ERRORRSPHER:ru nning out of buffer13 1 0non lr.F:Out of bufferRSPHER解决方案:根据CPU的数量,修改INCAR中NPAR,将NPAR=1修改成4 (或者2),问题得以解决。
错误3:WARNING: Sub-Space-Matrix is not hermitian in DAV 4 -4.681828688433112E-002Sub-Space-Matrix子空间矩阵、亚空间矩阵Hermitian厄米共轭警告:戴维森方法(DAV )中的子空间矩阵不是厄米共轭的。
解决方案:只需调整AMIX, BMIX 的值,把他们设置小一些。
一般采用其默认值,除非在电子迭代难以收敛的情况,才手动设置AMIX和BMIX等参数值。
经对Mixing方法的调试,通过将默认AMIX=0.4,修改成AMIX=0.2 (或0.3),问题得以解决。
Mixing 方法:IMIX=type ofmixi ng 混合、混频,AMIX=li near mixi ng parameter ,AMIN=mi ni mal mixi ngparameter,BMIX=cutoffwave vector for Kerker mixi ng scheme, AMIX_MAG=li near mixi ng parameter formagn etizati on,BMIX_MAG=cutoffwave vector for Kerker mixi ng scheme for mag, WC=weight factor for each stepin Broyde n mixi ng scheme,INIMIX=type ofinitial for each step in Broyden mixing scheme, MIXPRE=type of preconditioning inBroyde n mixing scheme,MAXMIX=maximu mn umber steps stored in Broyde n mixer.错误4:WARNING: Sub-Space-Matrix is not hermitian in DAV 1 -7.626640664998020E-003解决方案:在INCAR中加上IALG=Fast 已解决!(1QL、2QL已解决,3QL以上未解决)IALG=Fast (两种方法混用)IALGO=38 IALG=Normal 电子优化采用blockedDavidson 方法IALGO=48 IALG=Very_Fast 电子优化采用RMM-DIIS 算法错误5:ADVICE TO THIS USER RUNNING 'VASP/VAMP' (HEAR YOUR MASTER'SVOICE ...): You have a(more or less) 'small supercell' and for smaller cells it is recommended to usethe reciprocal-space projection scheme! The real space optimization is notefficient for small cells and it is also less accurate ... Therefore setLREAL=.FALSE. in the INCAR file解决方案:对于较小的晶胞(原子数小于20),设置LREAL=.FALSE.,计算结果比较精确。
而对于较大的晶胞,设置LREAL=Auto,这样计算速度比较快。
对于1QL 2QL 3QL 原子数分别为5、10、15,LREAL=.False.对于4QL 5QL 6QL 原子数分别为20、25、30,LREAL=Auto错误6:自旋轨道耦合计算时,静态和能带计算中出现的错误:ERROR: non colli near calculatio ns require that VASP is compiled withoutthe flag -DNGXhalf and -DNGZhalf错误:非线性计算需要编译过的VASP,VASP中不包含-DNGXhalf和-DNGZhalf解决方案:重新编译VASP。
don't forget that you may have to re-compile vasp without any of theprecompiler (CPP) flags set:-DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf , asn ecessary for non-colli near runs in generalfor non-colli near magn etism不要忘记如果你用的vasp不包含任何预编译程序命令-DNGXhalf, -DNGZhalf, -DwNGXhalf,-DwNGZhalf ,你必须重新编译vasp只有编译过,因为这些参数通常对于非线性磁性计算是必要的.错误7: SB-3QL 计算总是岀现这个错误VERY BAD NEWS! internal error in subroutine SGRCON:Fou nd some non-i nteger eleme nt in rotati on matrix 3解决办法:首先检查POSCAR是否有问题。