DNA甲基化研究方法的重大突破(原文)
植物DNA甲基化研究进展
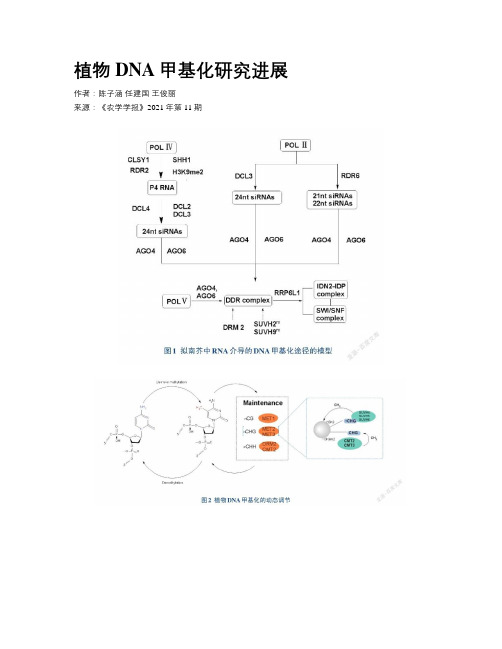
植物DNA甲基化研究进展作者:陈子涵任建国王俊丽来源:《农学学报》2021年第11期摘要:DNA甲基化是一種重要的表观遗传修饰,能够有效调控基因组稳定性。
为了了解DNA甲基化对植物生长发育的影响,本文归纳了近年来植物DNA甲基化的模式,总结了植物DNA甲基化的生物学功能,概括了DNA甲基化的研究方法,最后总结了植物DNA甲基化研究中存在的问题,并指明了研究方向,为后续植物基因组研究提供理论依据。
关键词:植物;DNA甲基化;表观遗传;修饰;生长发育;逆境胁迫;基因组;稳定性中图分类号:S184文献标志码:A论文编号:cjas2020-0152Research Advances on Plant DNA MethylationChen Zihan, Ren Jianguo, Wang Junli(School of Public Health, the key Laboratory of Enviromental Pollution Monitoring and Disease Control,Ministry of Education, Guizhou Medical University, Guiyang 550025, Guizhou, China)Abstract: DNA methylation is an important epigenetic modification that can effectively regulate genome stability. In order to understand the impact of DNA methylation on plant growth and development, this article summarizes plant DNA methylation patterns, concludes the physiological functions of plant DNA methylation, and reviews the research methods of DNA methylation. At last, this article sums up the problems in the study of plant DNA methylation and points out the research directions in the future, providing a theoretical basis for subsequent plant genome research.Keywords: Plants; DNA methylation; Epigenetic; Modification; Growth and Development; Adversity Stress; genome; stability0引言DNA甲基化(DNA methylation)是目前表观遗传学研究较为清晰的机制之一,广泛存在于生物界中,是真核细胞中最为常见的一种基因组修饰方式,它在调节基因组功能的同时不改变DNA的碱基序列。
线粒体DNA甲基化的研究进展

线粒体DNA甲基化的研究进展线粒体是细胞内的重要器官,它主要负责细胞内能量的生产,维持细胞正常的代谢活动。
线粒体DNA(mtDNA)在人类遗传和疾病中起着重要作用。
过去,人们通常认为线粒体DNA相对稳定,但近年来的研究表明,线粒体DNA也会受到一定的甲基化修饰。
线粒体DNA 的甲基化过程对细胞内能量代谢、氧化磷酸化和疾病发生发展有着重要的影响。
本文将对线粒体DNA甲基化的研究进展进行综述。
一、线粒体DNA甲基化的发现线粒体DNA甲基化是近年来的研究热点之一。
1997年,张玉宇等人首次报道了线粒体DNA存在甲基化修饰,证实了线粒体DNA也可以发生甲基化。
而早在1970年代,就有研究者观察到了线粒体DNA存在着甲基化的现象,但长期以来线粒体DNA甲基化研究一直处于较为初步的阶段。
二、线粒体DNA甲基化的研究方法对于线粒体DNA的甲基化研究,研究者主要采用了甲基化敏感的酶切法、甲基化特异性的PCR分析等方法。
也可以利用高通量测序技术对线粒体DNA进行全基因组甲基化分析,更全面地揭示线粒体DNA的甲基化水平和模式。
三、线粒体DNA甲基化与疾病关系近年来,越来越多的研究表明,线粒体DNA甲基化与多种疾病的发生和发展密切相关。
线粒体DNA甲基化异常与肿瘤的发生密切相关。
研究发现,在肿瘤组织中,线粒体DNA甲基化水平普遍显著升高,而且这种升高与肿瘤的发生、发展密切相关。
线粒体DNA甲基化与糖尿病、心血管疾病等多种疾病也有着密切的关系。
研究表明,这些疾病患者的线粒体DNA甲基化水平明显异常,对疾病的发展起到了重要的调控作用。
四、线粒体DNA甲基化与细胞代谢活动线粒体DNA甲基化还与细胞内的能量代谢活动密切相关。
研究表明,线粒体DNA甲基化水平的变化会对细胞内的氧化磷酸化、呼吸链和ATP合成等过程产生明显的影响。
线粒体DNA甲基化还可能会通过影响线粒体的功能,引起线粒体功能受损、能量代谢紊乱等,并最终导致多种疾病的发生。
DNA甲基化检测方法的研究进展_孙贝娜

12
生命科学仪器 2009 第 7 卷 /4 月刊
综述
扑获方法富集甲基化的 DNA 片段。 1.3.1 甲基化高密度芯片(CpG Islands microarray)
高密度的甲基化芯片是研究全基因甲基化状态的有 力工具,这类芯片可结合 DMH,MeDIP 或 MIRA 技术进 行研究获得高通量的数据结果。目前通用的高密度甲基 化芯片有 Agilent 公司及 Nimblegen 公司的商品化 CpG 岛 芯片[22],分别有人类,小鼠等,也可根据需要定制芯 片。Agilent 公司的芯片探针设计以 100-300bp 的平均间 隔覆盖所有 UCSC 注释的 CpG 岛和 RefSeq 数据库中所有 已研究清楚的约 17000 个转录本。每个甲基化芯片上都 集成有约 244,000 个 60-mer 的寡聚核苷酸探针,目前利 用原位合成的技术可实现在 1 " x 3 "的玻璃片基上灵 活地大规模地合成芯片探针。Nimblegne 公司的 CpG 岛 芯片密度更高,达 389,000 个探针[23]。甲基化芯片的整 个技术过程为:首先将基因组 DNA 超声打断成 400bp- 500bpDNA片段,将其加热变性并将变性后的单链DNA 样品分成两份:其中一份单链 DNA 样品加入抗 5'—甲 基化胞嘧啶核苷抗体,使用免疫磁珠法分离样品中甲 基化DNA片段的抗体复合物,样品中其余的非甲基化 DNA 片段被洗脱,纯化免疫共沉淀的 DNA 片段 (MeDIP);视需要可对 MeDIP 与 Input 样品进行扩增; 将MeDIP(Cy5)与Input(Cy3)样品分别进行荧光标记;标 记后的 MeDIP 与 Input 样品混合、变性,与 DNA 微阵 列芯片杂交;用高解析度芯片扫描仪检测杂交信号; 最后对杂交结果进行数据提取、标准化、峰值分析、报 告。甲基化高密度芯片可灵活进行甲基化研究,得到 高灵敏度、高特异性、高重复性的结果。目前拥有 的众多芯片专利技术中,每个芯片探针都经反复实验 筛选优化而来,芯片可使用双色或单色标记系统,双 色标记系统比单色标记系统得到灵敏度和精确度更 高、重复性更好的结果,既可以有效地检测出弱的、 低频 DNA 甲基化水平改变,又可以在同一张芯片上直 接比较不同样本的差异。Agilent 公司还专门设计了 ChIP Analytics Software 软件处理甲基化芯片数据[20], 保证研究者更好检测出 DNA 甲基化事件并降低错判 率。该软件还有可视化数据浏览器功能,研究者能 方便的将数据定位到基因组上,结合各种已有的基因 组注释进行研究。 1.3.2 差异甲基化杂交(Differential Methylation Hybridization, DMH)
《DNA甲基化调控印记及转录起始位点》范文

《DNA甲基化调控印记及转录起始位点》篇一一、引言DNA甲基化是生物学领域中的一个重要现象,对基因表达起着关键性作用。
其重要性体现在生命过程中的印记机制及转录起始位点的调控中。
印记现象为哺乳动物中的一种遗传机制,其作用在于确保父本和母本遗传信息的正确表达。
而转录起始位点则是基因表达的第一步,它直接关系到基因的激活和表达。
因此,本文将重点探讨DNA甲基化如何调控印记以及如何影响转录起始位点,进一步理解其在生物体内的功能和作用。
二、DNA甲基化及其功能DNA甲基化是生物学上的一种重要的基因表达调控方式。
具体而言,它是通过在DNA链的CpG二核苷酸上添加一个甲基基团来完成的。
这种修饰在许多生物过程中起着关键作用,包括基因沉默、X染色体失活、印记形成等。
三、DNA甲基化调控印记在哺乳动物中,印记是基因表达的一种重要机制,它决定了哪些基因由父本表达,哪些基因由母本表达。
这种机制在胚胎发育和生命维持中起着至关重要的作用。
DNA甲基化在印记的形成和维持中起着关键作用。
在胚胎发育的早期阶段,亲代DNA在遗传过程中会出现不均匀的甲化情况,形成了不同的甲基化印记。
这种甲化状态将直接影响胚胎内某些基因的开启和关闭状态,因此具有十分重要的功能。
这些不同的甲基化印记是由一系列的生物学因素共同作用而形成的,包括遗传因素和环境因素等。
四、DNA甲基化与转录起始位点的关系转录起始位点是基因表达的关键步骤之一,它决定了哪些基因将被激活并开始转录过程。
DNA甲基化在转录起始位点的调控中也起着重要作用。
研究表明,CpG二核苷酸的甲基化状态可以影响转录因子的结合和活性,从而影响转录起始位点的选择和基因的表达水平。
具体来说,当CpG二核苷酸被甲基化时,一些转录因子无法与其结合,导致基因的表达受到抑制;而当CpG二核苷酸未被甲基化时,这些转录因子则可以与其结合并启动基因的转录过程。
因此,DNA甲基化的状态可以决定哪些基因的转录起始位点被选择并激活。
线粒体DNA甲基化的研究进展

线粒体DNA甲基化的研究进展线粒体DNA甲基化是指线粒体DNA分子中的碱基上添加甲基基团的化学修饰。
它在线粒体功能调控、能量代谢和人类疾病中起着重要的作用。
本文将介绍线粒体DNA甲基化的研究进展。
线粒体DNA甲基化最早于2012年被发现。
研究人员利用高通量测序技术对小麦进行甲基化分析,发现线粒体DNA上存在着明显的甲基化信号。
随后的研究表明,线粒体DNA甲基化在多个物种中普遍存在。
通过对果蝇、线虫、鼠类等模式生物进行研究,科学家们发现线粒体DNA甲基化与线粒体功能密切相关。
在果蝇中,线粒体DNA甲基化水平的变化与线粒体呼吸链的功能紧密相关。
线粒体DNA甲基化是通过DNA甲基转移酶进行的。
具体来说,线粒体DNA甲基转移酶将甲基基团转移给线粒体DNA上的胞嘧啶(C)。
通过对线粒体DNA甲基化酶的功能屏蔽实验,研究人员发现该酶对线粒体功能的维持起着重要作用。
线粒体DNA甲基化在老化、肿瘤和心脏病等疾病中的调控也是研究热点。
研究人员发现,在老龄小鼠中线粒体DNA甲基化水平降低,而通过过表达线粒体DNA甲基化酶可以延缓老化进程。
线粒体DNA甲基化是由线粒体基因组自身进行的还是受到核基因组的调控仍然是一个有争议的问题。
一些研究表明,线粒体基因组存在独立于核基因组的甲基化修饰机制。
但另一些实验结果则表明,核基因组对线粒体DNA甲基化有重要的调控作用。
目前,尚不清楚线粒体DNA甲基化的修饰机制。
未来的研究方向包括进一步探究线粒体DNA甲基化与线粒体功能之间的关系,寻找新的线粒体DNA甲基化酶以及研究线粒体DNA甲基化在疾病中的作用机制。
对线粒体DNA甲基化的定量和定位等技术的开发也是重要的研究领域。
线粒体DNA甲基化在线粒体功能调控和人类疾病中具有重要作用。
随着研究的深入,我们对线粒体DNA甲基化的认识将更加全面,有望为相关疾病的预防和治疗提供新的思路和方法。
DNA甲基化检测的序曲——亚硫酸氢盐转化(转自生物通)

DNA甲基化检测的序曲——亚硫酸氢盐转化(转⾃⽣物通)检测DNA甲基化的⽅法有很多种,在众多⽅法中,研究⼈员常常会使⽤到⼀项技术,那就是亚硫酸氢盐转化。
DNA的亚硫酸氢盐处理将未甲基化的胞嘧啶转化成尿嘧啶,⽽甲基化的胞嘧啶保持不变。
随后⽤测序、定量PCR和芯⽚等分析来⽐较处理和未处理DNA的序列,就能确定哪些碱基是甲基化的。
尽管在理论上很简单,但亚硫酸氢盐处理真正要做起来却不那么容易。
如果你问⼀问那些做过的⼈,他们⼋成会告诉你不好做。
⾸先是回收率不⾼,会损失⼤量DNA,有时甚⾄达到50%。
其次是转化效率,有时是未能彻底转化,有时则是转化过了头。
此外,还要⾯临DNA的降解。
因为亚硫酸氢盐处理的剧烈条件,DNA会⽚段化,从⽽降低了PCR及后续分析技术的灵敏度。
⽬前市场上有多个⼚家提供亚硫酸氢盐转化试剂盒,包括QIAGEN、默克密理博、Sigma-Aldrich、Zymo、Life Technologies等。
在选购时,我们应注意关键的⼏点。
⾸先,确保所有的未甲基化CpG位点都能完全转化,才能获得可靠的结果。
另外,我们还要关⼼DNA是否能够⾼效回收。
⽅法不同,实验所需的时间也有差异。
传统的亚硫酸氢盐转化需要过夜孵育,⽽最新推出的试剂盒往往只需要1-3⼩时。
对于新⼿来说,最好还是买完整的试剂盒。
试剂盒中的试剂和操作步骤经过优化,容易得到更加⼀致的结果。
有些试剂盒中还带有对照。
有些⼈可能觉得对照可有可⽆,然⽽⼀旦出了问题,它的重要性就凸显了。
默克密理博——CpGenome™ Turbo Bisulfite Modification Kit这款极速重亚硫酸盐处理试剂盒能为你带来飞⼀般的感觉,让你体验极为快速的重亚硫酸盐转化。
使⽤这⼀款极速重亚硫酸盐处理试剂盒,从DNA样品处理到转化后的⾼纯DNA只需90分钟。
它的秘密就在于⼀款受专利保护的处理试剂,能在很短时间内将未甲基化的胞嘧啶彻底转化。
同时这种试剂对DNA的伤害⼜极⼩,不像通常的⾼盐、⾼温以及低pH等极端条件会导致DNA⾼度⽚段化。
DNA甲基化在肺癌早期诊断中的研究进展
健康域公卫从全世界范围来看,癌症死亡人数中占比最大的当数肺癌,根据GLOBOCAN(全球癌症流行病学的数据库)2020年数据显示,新增肺癌患者高达220万例(占癌症患者总数的11.4%),死于肺癌的人数高达179万例(占癌症总死亡人数的18.0%)。
采用低剂量计算机断层扫描(LDCT)对肺癌患者进行初步筛查能够提高肺癌的诊断率,从而提高肺癌患者生存率。
但是LDCT的灵敏度高而特异性低容易造成误诊,因此通过LDCT检测结果异常的患者需完善其他检查以进一步确认。
因此,仍需寻找简便易行、灵敏度和特异性好的无创手段对肺癌进行早期诊断[1]。
由于DNA甲基化与人类发育和肿瘤疾病密切相关,它是目前研究最透彻、最广泛的表观遗传学的现象之一。
DNA甲基化往往在肿瘤发病的早期甚至癌前病变时就已经发生,因此DNA甲基化的异常改变或可成为用于肺癌早期诊断的生物标记物。
为此本文主要就肺组织、外周血、肺泡灌洗液等标本当中DNA甲基化检测与肺癌早期诊断的研究进展进行综述。
1DNA甲基化的定义DNA甲基化是一类可稳定遗传的表观修饰,其参与了个体发育及细胞分化等基本生物学过程。
DNA甲基化是指在DNA甲基化转移酶的作用下,把甲基从通用甲基供体S-腺苷-L-甲硫氨酸添加到胞嘧啶的5-碳位置的一种化学修饰现象。
这种方式能够通过改变DNA的稳定性、DNA和蛋白质的互作方式以及染色质结构来调控靶向基因的表达。
多项研究表明,DNA的异常甲基化主要表现为全基因组的低甲基化状态和启动子CpG岛的高甲基化状态。
2DNA甲基化与肺癌肺癌的发生发展涉及一系列异常的表观遗传变化。
DNA甲基化作为表观遗传的重要修饰手段之一与癌症的发病有密切关系,是正常细胞癌变时最早发生的事件。
在癌变的早期阶段,不仅肿瘤细胞,甚至在非肿瘤细胞中也发生了异常的DNA甲基化。
抑癌基因失活的原因除了认为与碱基的突变和缺失有关外,另一个重要的原因被认为是其启动子区的高度甲基化。
研究DNA甲基化的分子生物学方法与技术
研究DNA甲基化的分子生物学方法与技术DNA甲基化是一种重要的表观遗传修饰形式,它在调控基因表达、细胞分化、肿瘤发生等方面发挥着关键作用。
针对DNA甲基化的研究需要运用到一系列的分子生物学方法和技术。
本文将就这些方法和技术进行讨论。
1. 甲基化特异性酶切实验DNA甲基化以CpG二核苷酸为主要靶点,可以运用甲基化特异性酶切酶(如MspI、HpaII等)进行评测。
具体操作流程是将DNA样品与相应的酶进行反应,再通过聚丙烯酰胺凝胶电泳等方法,分析产物的变化。
该方法具有简单、快速的优点,但由于CpG二核苷酸在基因组中分布不均衡,因此对于一些区域的甲基化评估有局限性。
2. 甲基化聚合酶链式反应(PCR)PCR技术已经成为分子生物学领域中不可或缺的一项技术手段。
运用甲基化特异性的PCR引物,通过PCR扩增后,CpG位点甲基化和非甲基化的两种细胞群体,可以进一步分析甲基化位点的变化。
该方法简单易行,可以进行大规模的样本检测。
3. 甲基化微阵列芯片(MeDIP)MeDIP是利用甲基化特异性抗体捕捉甲基化DNA序列,以实现全基因组水平的甲基化检测。
MeDIP是一种高通量测试方法,通常应用于发现肿瘤标志基因,评估细胞分化阶段等方面的研究。
4. 亚甲基化测序(TAB-seq)与MeDIP不同,TAB-seq针对亚甲基化的修饰进行评测。
亚甲基化是一种最新发现的甲基化形式,它的存在状态与基因的关闭和启动有着密切的关系。
运用TAB-seq技术,可以更加准确地评估亚甲基化的状态,有助于进一步探究亚甲基化在细胞分化、基因表达等方面的功能。
5. SMRT测序技术SMRT测序技术是一种全新的单分子测序技术,它的实现可以对DNA甲基化进行直接检测。
SMRT测序技术通过第三代测序技术、联合甲基化特异性的转录因子、氢氧化钠等多种方法,对DNA甲基化进行深入地评测。
与其他技术相比,SMRT可以不受区域分布不平衡的影响,可以更准确地评估DNA甲基化的状态。
《蛋白磷酸酶1及其DNA甲基化在低氧预适应小鼠海马和神经细胞中的变化情况》
《蛋白磷酸酶1及其DNA甲基化在低氧预适应小鼠海马和神经细胞中的变化情况》一、引言近年来,生物医学研究在深入探索生理及病理过程上取得了重大突破。
其中,蛋白磷酸酶1(PP1)及其DNA甲基化在低氧预适应过程中的作用,引起了科研人员的广泛关注。
本文将就这一主题展开讨论,分析PP1及其DNA甲基化在低氧预适应小鼠海马和神经细胞中的变化情况。
二、蛋白磷酸酶1(PP1)及其作用蛋白磷酸酶1(PP1)是一种在多种细胞中发挥重要作用的丝氨酸/苏氨酸磷酸酶。
它通过去磷酸化过程参与调节各种细胞内过程,包括神经传递、细胞凋亡和细胞生长等。
PP1在神经细胞中的关键作用表现在维持神经元的稳定性和神经网络的可塑性。
三、低氧预适应及其对小鼠海马和神经细胞的影响低氧预适应是一种生理过程,通过模拟低氧环境来提高机体对缺氧的耐受性。
低氧预适应能刺激机体内多种生理生化反应,其中对小鼠海马和神经细胞的影响尤为显著。
海马区是记忆和情感的重要部位,而神经细胞则对维持正常神经功能起着至关重要的作用。
四、蛋白磷酸酶1(PP1)在低氧预适应过程中的变化在低氧预适应过程中,PP1的表达和活性会发生变化。
具体来说,低氧环境会刺激PP1的表达增加,以应对缺氧带来的压力。
这种变化有助于维持神经细胞的稳定性和功能。
然而,这一过程的机制尚未完全明确,需要进一步的研究来揭示。
五、DNA甲基化在低氧预适应过程中的作用DNA甲基化是一种重要的表观遗传学机制,通过改变基因的表达而不改变基因序列来实现对生物过程的调控。
在低氧预适应过程中,DNA甲基化会发生变化,影响PP1和其他相关基因的表达。
这些变化可能对神经细胞的生存、分化和功能产生重要影响。
六、蛋白磷酸酶1(PP1)与DNA甲基化在低氧预适应小鼠海马和神经细胞中的变化情况在低氧预适应过程中,PP1的表达和活性与DNA甲基化之间存在密切的关联。
一方面,PP1的改变可能影响DNA甲基化的模式和程度;另一方面,DNA甲基化的变化也可能影响PP1的表达和活性。
《DNA甲基化调控印记及转录起始位点》范文
《DNA甲基化调控印记及转录起始位点》篇一一、引言生命中一个核心的过程就是遗传信息的复制与表达,其精确性与生物个体的生长和功能维护息息相关。
在遗传信息的表达过程中,DNA甲基化作为一种重要的表观遗传学机制,扮演着至关重要的角色。
本文将重点探讨DNA甲基化在调控印记及转录起始位点中的作用。
二、DNA甲基化的基本概念DNA甲基化是一种在DNA序列上添加甲基基团的过程,主要发生在CpG二核苷酸序列的胞嘧啶上。
在正常的人类细胞中,甲基化常位于特定的区域,这些区域包括印迹区域、CpG岛和调控元件等。
在许多情况下,甲基化影响的是基因的活性、染色体的稳定性以及遗传信息的传递。
三、DNA甲基化与印记调控印迹是一种遗传机制,使得父本和母本的等位基因表现出不同的表达模式。
印迹通常涉及母本和父本基因的特定基因沉默或表达增强,对于胎儿发育和生命维持具有重要作用。
在印迹的调控过程中,DNA甲基化起着关键作用。
在胚胎发育过程中,母本和父本的基因组会经历不同的甲基化模式。
这种差异化的甲基化模式可以导致某些基因的沉默或激活,从而影响印迹基因的表达。
此外,在发育过程中,某些基因的甲基化状态会随着时间和环境的变化而改变,这进一步强调了DNA甲基化在印迹调控中的重要性。
四、DNA甲基化与转录起始位点转录是基因表达的首个步骤,其过程始于转录起始位点。
在这个过程中,DNA甲基化能够影响转录因子的结合以及RNA聚合酶的活性,从而影响转录起始的效率和精确性。
具体来说,特定的甲基化模式可能会抑制或激活某些转录因子,进而影响基因的表达水平。
五、研究进展与展望近年来,随着表观遗传学研究的深入,DNA甲基化的作用得到了越来越多的关注。
许多研究表明,DNA甲基化与多种疾病的发生和发展密切相关,包括癌症、神经性疾病等。
因此,进一步研究DNA甲基化的作用机制以及其在各种生物过程中的具体应用具有重要意义。
未来的研究应继续深入探索DNA甲基化与印迹和转录起始位点之间的相互关系,以期更好地理解其在遗传信息复制和表达过程中的作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
10.1101/gr.136242.111Access the most recent version at doi: 2012 22: 1139-1143 originally published online March 30, 2012Genome Res.Andrew Adey and Jay ShenduresequencingUltra-low-input, tagmentation-based whole-genome bisulfiteMaterial Supplemental/content/suppl/2012/03/30/gr.136242.111.DC1.html References/content/22/6/1139.full.html#ref-list-1This article cites 15 articles, 5 of which can be accessed free at:License Commons Creative ./licenses/by-nc/3.0/described at as a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), ). After six months, it is available under /site/misc/terms.xhtml for the first six months after the full-issue publication date (seeThis article is distributed exclusively by Cold Spring Harbor Laboratory Press serviceEmail alertingclick here top right corner of the article or Receive free email alerts when new articles cite this article - sign up in the box at the/subscriptions go to: Genome Research To subscribe to © 2012, Published by Cold Spring Harbor Laboratory PressMethodUltra-low-input,tagmentation-based whole-genome bisulfite sequencingAndrew Adey and Jay Shendure1Department of Genome Sciences,University of Washington,Seattle,Washington98195,USAWe have adapted transposase-based in vitro shotgun library construction(‘‘tagmentation’’)for whole-genome bisulfite sequencing.This method,Tn5mC-seq,enables a>100-fold reduction in starting material relative to conventional protocols,such that we generate highly complex bisulfite sequencing libraries from as little as10ng of input DNA,and ample useful sequences from1ng of input DNA.We demonstrate Tn5mC-seq by sequencing the methylome of a human lymphoblastoid cell line to~8.63high-quality coverage of each strand.[Supplemental material is available for this article.]DNA methylation is a widespread epigenetic modification that plays a pivotal role in the regulation of the genomes of diverse organisms.The most prevalent and widely studied form of DNA methylation in mammalian genomes occurs at the five carbon position of cytosine residues,usually in the context of the CpG dinucleotide.Microarrays,and more recently massively parallel sequencing,have enabled the interrogation of cytosine methyl-ation(5mC)on a genome-wide scale(Zilberman and Henikoff 2007).However,the in vivo study of DNA methylation and other epigenetic marks,e.g.,in specific cell types or anatomical struc-tures,is sharply limited by the relatively high amount of input material required for contemporary protocols.Methods for genome-scale interrogation of methylation pat-terns include several that are preceded by the enrichment of de-fined subsets of the genome(Meissner et al.2005;Down et al. 2008;Deng et al.2009),e.g.,reduced representation bisulfite se-quencing(RRBS)(Meissner et al.2005)and anti-methylcytosine DNA immunoprecipitation followed by sequencing(MeDIP-seq) (Down et al.2008).An advantage of such methods is that they can be performed with limited quantities of starting DNA(Gu et al. 2011).However,they are constrained in that they are not truly comprehensive.For example,the digestion-based RRBS method interrogates only;12%of CpGs,primarily in CpG islands(Harris et al.2010),with poor coverage of methylation in gene bodies(Ball et al.2009)and elsewhere.Furthermore,RRBS does not target cy-tosines in the CHG or CHH(H=A,C,T)contexts,which have been shown to be methylated at elevated levels in the early stages of mammalian development(Lister et al.2009).While a small pro-portion of non-CpG methylation sites can be observed using RRBS, they are restricted to regions within or highly proximal to CpG-islands(Ziller et al.2011).The most comprehensive,highest resolution method for detecting5mC is whole-genome bisulfite sequencing(WGBS) (Cokus et al.2008;Lister et al.2009;Harris et al.2010).Treatment of genomic DNA with sodium bisulfite chemically deaminates cytosines much more rapidly than5mC,preferentially converting them to uracils(Clark et al.1994).With massively parallel se-quencing,these can be detected on a genome-wide scale at single-base-pair resolution.This approach has revealed complex and unexpected methylation patterns and variation,particularly in the CHG and CHH contexts.Furthermore,as the costs of massively parallel sequencing continue to plummet,WGBS is increasingly affordable.However,a key limitation of WGBS is that the current protocols for library construction are based on ligation chemistry and call for5m g of genomic DNA as input(Cokus et al.2008;Lister et al.2009;Li et al.2010)which is essentially prohibitive for many samples obtained in vivo.We recently characterized a transposase-based in vitro shot-gun library construction method(‘‘tagmentation’’)that allows for construction of sequencing libraries from greatly reduced amounts of DNA(Fig.1A;Adey et al.2010).Briefly,the method utilizes a hyperactive derivative of the Tn5transposase loaded with dis-continuous synthetic oligonucleotides to simultaneously fragment and append adaptors to genomic DNA.The resulting products are subjected to PCR amplification followed by high-throughput se-quencing.The increased efficiency of genomic DNA conversion to viable amplicons and the greatly reduced number of steps allow the construction of low-bias,highly complex libraries from<50ng of genomic DNA.Here we describe a modified approach,which we call Tn5mC-seq,that retains the advantages of transposase-based library preparation in the context of WGBS.Because the target of the transposition reaction is double-stranded DNA,whereas bisulfite treatment yields single-stranded DNA,the method was extensively modified such that the tagmentation reaction could take place prior to bisulfite treatment(Fig.1B).First,the adaptors to be in-corporated were methylated at all cytosine residues to maintain cytosine identity during bisulfite treatment,with the exception of the19-bp transposase recognition sequence(in order to minimize differential binding during transposome assembly).Second,an oligonucleotide replacement scheme(Supplemental Fig.S1B; Grunenwald et al.2011;Gertz et al.2012)was utilized to ensure that each strand would have adaptors covalently attached to both ends of the molecule.Specifically,this entails initial transposition with a single adaptor in which the double-stranded transposase recognition sequence is truncated to16bp(T m=36°C),thereby facilitating its post-incorporation removal by denaturation.A second adaptor is then annealed and the gap repaired,resulting in each strand being covalently flanked by both a39and59adaptor. The fragmented,adapted,double-stranded genomic DNA is then subjected to standard bisulfite treatment for the conversion of unmethylated cytosine to uracil.Degradation during the conver-sion process likely remains a primary source of loss,but the in-1Corresponding author.E-mail shendure@.Article published online before print.Article,supplemental material,and publi-cation date are at /cgi/doi/10.1101/gr.136242.111.22:1139–1143Ó2012,Published by Cold Spring Harbor Laboratory Press;ISSN1088-9051/12; Genome Research1139creased efficiency of the prior steps and the lack of gel-based sizeselection result in an overall increase in the fraction of DNA that isconverted,PCR-amplified,and sequenced.Results Ultra-low-input transposase-based WGBS library performance We applied Tn5mC-seq to sequence the methylome of a lympho-blastoid cell line (GM20847)using libraries constructed from 1–200ng of input genomic DNA.Each library was barcoded during PCRamplification and subjected to either a spike-in (5%)or majority(80%–90%)of a lane of sequencing on an Illumina HiSeq2000(paired-end 100bp [PE100];v2chemistry with custom sequencingprimers).These data are summarized in Table 1and SupplementalFigure S2.In addition,several PCR conditions were investigated tooptimize amplification uniformity (Sup-plemental Fig.S3),as well as a modified protocol (Tn5mC-seq 1.1)(Supplemen-tal Figs.S1D,S4)that eliminates the need for custom sequencing primers and may increase library construction efficiency.Reads were aligned to an in silico con-verted hg19(GRC37)to both the top (C !T)and bottom (G !A)strands using BWA (Li and Durbin 2009)followed by read trimming of unmapped reads and secondary alignment using the same pa-rameters.Unaligned reads typically con-sisted of low-quality artifacts that likely arose during amplification due to the re-duced base complexity of bisulfite con-verted amplicons.For each library constructed using $10ng of genomic DNA,over 100million aligned reads were obtained (60%–75%of total filtered reads;see Methods)of high complexity (90%–97%nondupli-cates).Despite the significantly reduced performance of libraries prepared from 1ng,;12million reads were still aligned and the library was of reasonable com-plexity (78%nonduplicates).Post-align-ment reads were merged and quality fil-tered for a total of 51.7Gb of aligned,unique sequence.The average read depth was 8.63per strand with >96%of CpG and >98%of non-CpG cytosines covered genome-wide (Fig.1C;SupplementalFig.S2).Because unmethylated nucleo-tides are incorporated during the gap-re-pair step (first 9bp of the second read and last 9bp before the adaptor as determined by insert size on the first read),the gap-repair regions must be excluded from methylation analysis.However,these bases also serve as an internal control for the conversion rate of the bisulfite treat-ment.We found this to be >99%for all li-braries,and this was independently con-firmed using unmethylated lambda DNA spike-ins to two libraries.For comparison,ligation chemistry–based libraries were constructed using 1000,100,and 10ng of GM20847DNA of the same isolation as the batch used for Tn5mC-seq.These libraries were prepared following the protocols outlined by Lister et al.(2009)with the exception of PCR,which was performed using Kapa Robust dueto its higher efficiency over other polymerase choices (Supple-mental Fig.S3)During amplification,the 100and 10ng prepara-tions did not show significant amplification above a negativecontrol background and were not carried through to sequencing,precluding a comparison of Tn5mC-seq and ligation chemistry–based library construction with identical inputs (a 1000ng Tn5mC-seq preparation was also not feasible due to the dilute concentra-tions of the commercially available transposase,which would result in a reduced density of transposition events on a high input mass).Post-alignment,the 1000ng ligation chemistry–based library provided slightly more uniform coverage than Tn5mC-seq1.1Figure 1.The Tn5mC-seq method and resulting methylation profiles.(A )Tagmentation-based DNA-seq library construction.Genomic DNA is attacked by transposase homodimers loaded with synthetic,discontinuous oligos (yellow,purple)that allow for fragmentation and adaptor incorporation in a single step.Subsequent PCR appends outer flowcell-compatible primers (pink,green).(B )Tn5mC-seq libraryconstruction.Loaded transposase attacks genomic DNA with a single methylated adaptor (yellow).Anoligo-replacement approach anneals a second methylated adaptor (purple),which is then subject togap-repair.Bisulfite treatment then converts unmethylated cytosine to uracil (orange)followed by PCRto append outer flowcell-compatible primers (pink,green).Methylation is represented as black lolli-pops.(C )Coverage of cytosine positions genome-wide.More than 96%of Cs in all three contexts are covered at least once.Slight decrease in CpG coverage is due to reduced read alignment ability atregions with a high density of methylation.(D )Normalized methylated cytosine over total cytosinepositions in 10-kb windows across chromosome 12(blue and purple,left axis),and normalized meth-ylated CpG over total CpG in 100-kb windows across chromosome 12(green,right axis).(E )Normalized methylated CpG over total CpG residues at annotated genic loci.Promoter is defined as 2-kb region upstream of TSS.(F )Elevated CpG methylation levels in gene body (intron,exon)compared to inter-genic regions.1140Genome ResearchAdey and Shendure(Supplemental Fig.S1D)libraries constructed from10ng,partic-ularly at the lower CpG densities that represent the majority of the genome(Supplemental Fig.S5A).Comparable uniformity was also observed with respect to G+C content as well as for tetramer/ pentamer sequence contexts(Supplemental Fig.S5B,C).We also compared the methylation levels of CpGs well-covered by se-quencing of libraries corresponding to both methods,and ob-served good agreement at positions with53or greater coverage (r2=0.55)as well as103or greater coverage(r2=0.82)(Supple-mental Fig.S5D).Lymphoblastoid cell line methylationWe were able to detect;46million5mC positions(1%FDR;see Methods),accounting for4.2%of total cytosines with coverage. The majority of methylation observed was in the CpG context (97.1%),and the global CpG methylation level was69.1%.This level is similar to that of the fetal fibroblast cell line IMR90se-quenced by Lister et al.(2009;67.7%)and is consistent with the observation that CpG methylation levels are reduced in differen-tiated cell types.Additionally,CHG and CHH methylation levels were substantially lower than in ES cells,at0.36%and0.37%,re-spectively,again consistent with the differentiated cell type.On the chromosome scale,the methylation density correlated with banding patterns and increasing levels were observed extending distally through subtelomeric regions(Fig.1D).An analysis of functionally annotated genic regions revealed a sharp decrease in CpG methylation through the promoter region followed by a mi-nor increase in the59UTR and then elevated levels of methylation throughout the gene body,particularly at introns(Fig.1E,F), consistent with previously described CpG methylation profiles (Lister et al.2009).DiscussionWe developed Tn5mC-seq as a novel method for rapidly preparing complex,shotgun bisulfite sequencing libraries for WGBS.In brief, the method utilizes a hyperactive Tn5transposase derivative to fragment genomic DNA and append adaptors in a single step,as previously characterized for the construction of DNA-seq libraries (Adey et al.2010).In order for library molecules to withstand bi-sulfite treatment,the adaptors are methylated at all cytosine resi-dues,and an oligonucleotide replacement strategy is employed to make each single-strand covalently flanked by adaptors.The high efficiency of the transposase and overall reduction in loss-associated steps permits construction of high-quality bisulfite se-quencing libraries from as little as10ng that are comparable to ligation chemistry–based libraries generated from1003more DNA,as well as useful sequence from1ng of input DNA.Addi-tionally,the increased efficiency of transposase-mediated library construction may allow for preparation of WGBS libraries from poor-quality or degraded DNA samples.Our results illustrate how derivatives of the transposase-based method for DNA-seq library preparation can enable key applica-tions of next-generation sequencing where its advantages are perhaps even more relevant.The ability to generate such libraries from very low amounts of input genomic DNA substantially im-proves the practicality of whole methylome sequencing and re-moves a key advantage of less encompassing methods such as RRBS (Meissner et al.2005;Harris et al.2010).Specifically,low-input WGBS with Tn5mC-seq may make possible the comprehensive interrogation of methylation in many contexts where DNA quantity is a bottleneck,e.g.,developing anatomical structures, microdissected tissues,or pathologies such as cancer,where the epigenetic landscape is of interest but tissue quantity limits high-resolution WGBS.MethodsTn5mC-seq library construction and sequencing Transposome complexes were generated by incubating2.5m L of 10m M Tn5mC-A1(Tn5mC-A1top:59-GAT[5mC]TA[5mC]A[5mC] G[5mC][5mC]T[5mC][5mC][5mC]T[5mC]G[5mC]G[5mC] [5mC]AT[5mC]AGAGATGTGTATAAGAGACAG-39,IDT,an-nealed to Tn5mC-A1bot:59-[Phos]-CTGTCTCTTATACACA-39,IDT, by incubating10m L of each oligo at100m M and80m L of EB [QIAGen]for2min at95°C and then cooling to room temperature at0.1°C/sec)with2.5m L100%glycerol and5m L Ez-Tn5transposase (Epicentre–Illumina)for20min at room temperature.Genomic DNA prepared from GM20847cell lines was used at respective input quantities with4m L Nextera HMW Buffer (Epicentre-Illumina),nuclease-free water(Ambion)to17.5m L and 2.5m L prepared Tn5mC transposomes(regardless of the quantity of DNA used).Reactions were incubated for9min at55°C in a ther-mocycler followed by SPRI bead cleanup(AMPure)using36m L of beads and the recommended protocol with elution in14m L nuclease-free water(Ambion).Adaptor2annealing was then carried out by adding2m L of103Ampligase Reaction Buffer(Epicentre-Illumina),2m L103dNTPs(2.5mM each;Invitrogen),and2m L 10m M Tn5mC-A2top(59-/5Phos/CTGTCTCTTATACACATCT[5mC] TGAG[5mC]GGG[5mC]TGG[5mC]AAGG[5mC]AGA[5mC] [5mC]GAT[5mC]-39;IDT)to each reaction and incubating for 2min at50°C followed by10min at45°C and cooling at0.1°C/sec to 37°C and subsequent incubation for10min.Gap-repair was then performed by adding3m L of Ampligase at5U/m L(Epicentre-Illu-mina)and1m L of either T4DNA Polymerase(Tn5mC libraries A-G, NEB)or Sulfolobus DNA Polymerase IV(Tn5mC libraries H-J,NEB) and additional incubation for30min at37°C.Reactions were then cleaned up using SPRI beads(AMPure)according to recommended protocol using36m L beads and elution in50m L nuclease-free water(Ambion).Bisulfite treatment was performed using an EZTable1.Summary of Tn5mC-seq libraries and sequencingName InputDNA(ng)PercentagealigningPercentageuniqueUniquealignedreadsMeaninsertsize(bp)Tn5mC-C2006893127,098,152198Tn5mC-D507590133,383,834254Tn5mC-E a1127611,181,960134Tn5mC-F a106595118,170,302168Tn5mC-G a50619787,294,793180Tn5mC-H1117812,393,357126Tn5mC-I b1062n/a29,546,077n/aTn5mC-J507195132,144,644196TOTAL651,213,119Raw reads were initially filtered for instrument valve failures at specificlocations of reads and then removal of reads containing over three Ns orextremely low-quality bases(phred score#2)in the first50bases.Alignment was then performed using BWA(Li and Durbin2009)to in silicoconverted top and bottom strand references of hg19(GRC37)followed bytrimming and realignment.Duplicate reads were identified and removedaccording to their start position and insert size.The percentage of post-filtering reads that align for each library is shown,as is the percentage ofthese that are nonduplicates.a Valve failures in read2resulted in extensive read trimming(50–70bp).b Complete valve failure on read2.Ultra-low-input whole-genome bisulfite sequencingGenome Research1141DNA Methylation Kit(Zymo)according to recommended pro-tocols with a14-h50°C incubation and10m L elution.Eluate was then used as the template for PCR using12.5m L Kapa2G Robust HotStart ReadyMix(Kapa Biosystems),1m L10m M Tn5mC-P1(59-AATGATACGGCGACCACCGAGATCTACACGCCTCCCTCGCG CCATCAG-39;IDT),1m L10m M barcoded P2(from Adey et al.2010), 0.15m L1003SYBR Green(Invitrogen),and0.35m L nuclease-free water(Ambion).Thermocycling was carried out on a BioRad Opticon Mini real-time machine with the following parameters:5 min at95°C;(15sec at95°C;15sec at62°C;40sec at72°C;Plate Read;10sec at72°C)399.Reactions were monitored and removed from thermocycler as soon as plateau was reached(12–15cycles).Sequencing was carried out using either a full or partial lane on an Illumina HiSeq2000using custom sequencing primers: read1,Tn5mC-R1(59-GCCTCCCTCGCGCCATCAGAGATGTGTATA AGAGATAG-39;IDT);index read,Tn5mC-Ix(59-TTGTTTTTTATATA TATTTCTGAGCGGGCTGGCAAGGC-39;IDT);and read2,Tn5mC-R2 (59-GCCTTGCCAGCCCGCTCAGAAATATATATAAAAAACAA-39;IDT). Read lengths were either single-read at36bp with a9-bp index (SE36,libraries A and B,not included in Table1)or101bp paired-end with a9-bp index(PE101,libraries C–J).Libraries were only sequenced on runs that did not have lanes containing Nextera li-braries as a precaution due to the similarity between sequencing primers.Tn5mC-Seq1.1library preparation(Supplemental Fig.1D) was carried out as previously described with several modifications: (1)Transposase recognition sequence reverse compliment is39 blocked to prevent nonspecific extension in final PCR.(2)Re-placement oligo is methylated through the region complementary to the transposase recognition sequence to maintain complexity during bisulfite conversion and allow the use of standard Nextera sequencing primers.(3)Replacement oligo is39blocked to prevent degradation by39!59exonuclease activity of gap-repair poly-merase(replacement oligo:Tn5mC1.1-A2top59-/5Phos/[5mC] TGT[5mC]T[5mC]TTATA[5mC]A[5mC]AT[5mC]T[5mC] TGAG[5mC]GGG[5mC]TGG[5mC]AAGG[5mC]AGA [5mC][5mC]GA[inv dT]-39,IDT;blocked transposase recognition sequence end:Tn5mC1.1-A1bot3block59-[Phos]-CTGTCTCTTA TACA[ddC]-39).Duplicate libraries were prepared from100ng, 10ng,and1ng of starting material and were subject to PCR am-plification using either Kapa HiFi U+Hot Start Ready Mix,or Kapa2G Robust Hot Start Ready Mix(Kapa Biosystems)and were sequenced on a single-end36-bp read plus a9-bp index read run on an Illu-mina GAIIx.Library characterization can be found in Supple-mental Figure5.Ligation chemistry WGBS library construction and sequencing We subjected1000,100,and10ng of genomic DNA prepared from GM18507cell lines to ligation chemistry–based library prepara-tion according to methods described by Lister et al.(2009)with several minor exceptions:(1)Bisulfite conversion was carried out using an EZ DNA Methylation Kit(Zymo),and(2)PCR was carried out using Kapa2G Robust Hot Start Ready Mix(Kapa Biosystems). The change in PCR enzyme was due to several unpublished ex-periments demonstrating a much higher efficiency with Kapa Robust as opposed to PfuTurbo Cx used according to the method described by Lister et.al.(2009).Sequencing was performed on an Illumina MiSeq instrument using a single-end100-bp sequence read run.Read filtering and alignmentThe hg19reference genome was first bisulfite-converted in silico for both the top(C changed to T,C2T)and bottom(G changed to A,G2A)strands.Prior to alignment,reads were filtered based on the run metrics,as several libraries were run on lanes in which instrument valve failures resulted in poor quality or reads con-sisting primarily of N bases.Filtering was carried out by first cal-culating the base compositions as well as mean base quality scores at each position in the read.Many of the lanes had significantly reduced quality scores at the start and/or end of the read and were globally trimmed to remove any start or end positions that had a mean phed score of less than or equal to10.The start and ends of the reads were additionally globally trimmed if a position within the first or last25bases of the read had a mean composition of10% Ns,which generally corresponded to the quality-based trimming. Additionally,reads that contained three or more Ns were also re-moved.It is important to note that the reduced qualities in the runs were‘‘flowcell-wide’’regardless of the library that was run and not isolated to Tn5mC-seq libraries.Subsequent runs for the Tn5mC-seq1.1and polymerase testing experiments did not suffer instrument failures,and no trimming of the reads was necessary. Next,reads were aligned to both the C2T and G2A strands using BWA with default parameters.Reads that aligned to both strands were removed.Read pairs in which neither aligned to either strand were then pulled and trimmed to76bp(except for SE36runs)and again aligned to both C2T and G2A strands.Duplicate reads(pairs sharing the same start positions for both reads1and2)were re-moved and complexity determined.Reads with an alignment score less than10were then filtered out prior to secondary analysis.Total fold coverage was calculated using the total bases aligned from unique reads over the total alignable bases of the genome(slightly below3Gb per strand).5mC callingMethylated cytosines were called using a binomial distribution as in the method described by Lister et al.(2009),whereby a proba-bility mass function is calculated for each methylation context (CpG,CHG,CHH)using the number of reads covering the position as the number of trials and reads maintaining cytosine status as successes with a probability of success based on the total error rates that were determined by the combined nonconversion rate and sequencing error rate.The total error rate was initially determined by unmethylated lambda DNA spike-ins;however,we found that the error rate estimation from the gap-repair portion of reads(as described in the main text)gave a more comprehensive estimate, which was slightly higher than that of the lambda estimate. Therefore to be conservative,we used the highest determined error rate at0.009.If the probability was below the value of M,where M3(number of total unmethylated CpG)<0.013(number of total methylated CpG),the position was called as being methylated, thus enforcing that no more than1%of positions would be due to the error rate.Data accessThe sequence data presented in this study have been submitted to the NCBI Sequence Read Archive(SRA)(http://www.ncbi.nlm. /sra)under accession no.SRP011746. AcknowledgmentsWe thank Cholie(Charlie)Lee for performing all sequencing runs and the Shendure laboratory for helpful discussions.We also thank Nick Caruccio,Haiying Gruenwald,Brad Baas,and Igor Goryshin from Epicentre(Illumina)for help and ideas regarding transposase-based library preparation,as well as Eric Van Der Walt and col-leagues at Kapa Biosystems for early access to reagents and pro-Adey and Shendure1142Genome Researchtocols for library amplification.A.A.is funded by an NSF Graduate Research Fellowship.This work was supported in part by the Lowell Milken Prostate Cancer Foundation Young Investigator Award(J.S.).Author contributions:A.A.performed experiments and data analysis.A.A.and J.S.designed experiments and wrote the manuscript. ReferencesAdey A,Morrison HG,Asan,Xun X,Kitzman JO,Turner EH,Stackhouse B, MacKenzie AP,Caruccio NC,Zhang X,et al.2010.Rapid,low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition.Genome Biol11:R119.doi:10.1186/gb-2010-11-12-r119.Ball MP,Li JB,Gao Y,Lee JH,LeProust EM,Park IH,Xie B,Daley GQ, Church GM.2009.Targeted and genome-scale strategies revealgene-body methylation signatures in human cells.Nat Biotechnol 27:361–368.Clark SJ,Harrison J,Paul CL,Frommer M.1994.High sensitivity mapping of methylated cytosines.Nucleic Acids Res22:2990–2997.Cokus SJ,Feng S,Zhang X,Chen Z,Merriman B,Haudenschild CD,Pradhan S,Nelson SF,Pellegrini M,Jacobsen SE.2008.Shotgun bisulphitesequencing of the Arabidopsis genome reveals DNA methylationpatterning.Nature452:215–219.Deng J,Shoemaker R,Xie B,Gore A,LeProust EM,Antosiewicz-Bourget J, Egli D,Maherali N,Park IH,Yu J,et al.2009.Targeted bisulfitesequencing reveals changes in DNA methylation associated withnuclear reprogramming.Nat Biotechnol27:353–360.Down TA,Rakyan VK,Turner DJ,Flicek P,Li H,Kulesha E,Graf S,Johnson N, Herrero J,Tomazou EM,et al.2008.A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis.NatBiotechnol26:779–785.Gertz J,Varley KE,Davis NS,Baas BJ,Goryshin IY,Vaidyanathan R,Kuersten S,Myers RM.2012.Transposase mediated construction of RNA-seqlibraries.Genome Res22:134–141.Grunenwald H,Baas B,Goryshin I,Zhang B,Adey A,Hu S,Shendure J, Caruccio N,Maffitt M.2011.Nextera PCR-free DNA library preparation for next-generation sequencing.(Poster presentation,AGBT2011).Gu H,Smith ZD,Bock C,Boyle P,Gnirke A,Meissner A.2011.Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling.Nat Protoc6:468–481.Harris RA,Wang T,Coarfa C,Nagarajan RP,Hong C,Downey SL,Johnson BE,Fouse SD,Delaney A,Zhao Y,et parison of sequencing-based methods to profile DNA methylation and identification ofmonoallelic epigenetic modifications.Nat Biotechnol28:1097–1105.Li H,Durbin R.2009.Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics25:1754–1760.Li Y,Zhu J,Tian G,Li N,Li Q,Ye M,Zheng H,Yu J,Wu H,Sun J,et al.2010.The DNA methylome of human peripheral blood mononuclear cells.PLoS Biol8:e1000533.doi:10.1371/journal.pbio.1000533.Lister R,Pelizzola M,Dowen RH,Hawkins RD,Hon G,Tonti-Filippini J,Nery JR,Lee L,Ye Z,Ngo QM,et al.2009.Human DNA methylomes at base resolution show widespread epigenomic differences.Nature462:315–322. Meissner A,Gnirke A,Bell GW,Ramsahoye B,Lander ES,Jaenisch R.2005.Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis.Nucleic Acids Res33:5868–5877. Zilberman D,Henikoff S.2007.Genome-wide analysis of DNA methylation patterns.Development134:3959–3965.Ziller MJ,Muller F,Liao J,Zhang Y,Gu H,Bock C,Boyle P,Epstein CB, Bernstein BE,Lengauer T,et al.2011.Genomic distribution and inter-sample variation of non-CpG methylation across human cell types.PLoS Genet7:e1002389.doi:10.1371/journal.pgen.1002389.Received December11,2011;accepted in revised form March29,2012.Ultra-low-input whole-genome bisulfite sequencingGenome Research1143。