残留溶剂测定法二部检验标准操作规程
09T-I638-01中华人民共和国药典(二部)制剂通则检验标准操作规程
——————————文件类别:技术标准 1/121.目的:建立《中华人民共和国药典》(二部)制剂通则检验标准操作规程,并按规程进行检验,保证检验操作规范化。
2.依据:2.1.《中华人民共和国药典》2010年版二部。
3.范围:适用于所有用《中华人民共和国药典》(二部)制剂通则测定的供试品。
4.责任:检验员、质量控制科主任、质量管理部经理对本规程负责。
5.正文:制剂通则5.1. 附录ⅠA 片剂。
5.1.1. 片剂系指药物与适宜的辅料混匀压制而成的圆片状或异形片状的固体制剂。
5.1.2. 片剂以口服普通片为主,另有含片、舌下片、口腔贴片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾片、缓释片、控释片与肠溶片等。
5.1.2.1. 含片:系指含于口腔中缓慢溶化产生持久局部或全身作用的片剂。
5.1.2.1.1. 含片中的药物应是易溶性的,主要起局部消炎、杀菌、收敛、止痛或局部麻醉作用。
5.1.2.1.2. 含片的溶化性照崩解时限检查法(附录Ⅹ A)检查,除另有规定外,10分钟内不应全部崩解或溶化。
5.1.2.2. 舌下片:系指置于舌下能迅速溶化,药物经舌下黏膜吸收发挥全身作用的片剂。
5.1.2.2.1. 舌下片中的药物与辅料应是易溶性的,主要适用于急症的治疗。
5.1.2.2.2. 舌下片中的药物与辅料应是易溶性的,主要适用于急症的治疗。
5.1.2.2.3. 舌下片照崩解时限检查法(附录Ⅹ A)检查,除另有规定外,应在5分钟内全部溶化。
5.1.2.3. 口腔贴片:系指粘贴于口腔,经黏膜吸收后起局部或全身作用的片剂。
5.1.2.3.1. 口腔贴片应进行溶出度或释放度检查。
5.1.2.4. 咀嚼片:系指于口腔中咀嚼后吞服的片剂。
5.1.2.4.1. 咀嚼片一般应选择甘露醇、山梨醇、蔗糖等水溶性辅料作填充剂和黏合剂。
咀嚼片的硬度应适宜。
5.1.2.5. 分散片:系指在水中能迅速崩解并均匀分散的片剂。
5.1.2.5.1. 分散片中的药物应是难溶性的。
乙交酯丙交酯共聚物(5050)(供注射用)成品检验操作规程

一、目的:制订详尽的工作程序,规范检验操作,保证检验数据的准确性。
二、范围:适用于乙交酯丙交酯共聚物(5050)(供注射用)检验的标准操作程序。
三、职责:1、检验员:严格按操作规程操作,认真、及时、准确地填写检验记录;2、化验室负责人:监督检查检验员执行本操作规程。
四、内容:1【性状】1.1本品为白色至淡黄色粉末或颗粒,几乎无臭。
1.2本品在三氯甲烷、二氯甲烷、丙酮、二甲基甲酰胺中易溶,在乙酸乙酯中微溶,在水、乙醇、乙醚中不溶。
2【鉴别】取本品0.5g,精密称定,置100ml量瓶中,加三氯甲烷70ml,超声至完全溶解,冷却至室温后,加三氯甲烷稀释至刻度,摇匀。
取上述溶液,(见EK/SOP-QC7040红外分光光度法检查操作规程)测定样品红外光谱。
3、【检查】3.1 酸度:取本品适量,研细,加水超声10分钟,分散成约2.0mg/ml的混悬液,过滤,取续滤液,依法测定(见EK/SOP-QC7003 PH测定操作规程),pH应为5.0~7.0。
3.2溶液的澄清度:取本品0.5g,加二氯甲烷25ml使溶解,依法检查(见EK/SOP-QC7011溶液澄清度与颜色检查操作规程),溶液应澄清。
3.3 乙交酯和丙交酯取乙酸丁酯适量,精密称定,加二氯甲烷溶解,并制成1ml中约含0.125mg的溶液,作为内标溶液;取本品约0.1g,精密称定,置10ml量瓶中,加内标溶液2ml,取二氯甲烷溶解,并稀释至刻度,摇匀,作为供试品溶液;另分别取乙交酯、丙交酯适量,精密加入内标溶液适量,用二氯甲烷溶解并制成每1ml中约含乙交酯50μg、丙交酯100μg、乙酸丁酯25μg的溶液,作为对照溶液。
照气相色谱法(见EK/SOP-QC7039气相色谱法检测操作规程)测定。
以5%苯基-甲基聚硅氧烷(或极性相近)为固定液的色谱柱,柱温为135℃,进样口温度为250℃,检测器温度为300℃。
取供试品溶液与对照溶液各3μl,分别注入气相色谱仪,按内标法以峰面积计算,含丙交酯不得过1.5%,乙交酯不得过0.5%。
GMP认证全套文件资料55-溶出度测定法标准操作规程

- 溶出度测定法标准操作规程目的:建立溶出度测定法标准操作规程。
适用范围:溶出度测定。
责任:质检员实施本操作规程,检验室主任负责监督本规程正确执行。
程序:1.简述1.1溶出度(中国药典2000年版二部附录X C)是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。
它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。
1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出的量。
1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。
1.4中国药典2000年版收载三种测定方法,第一法转篮法第二法桨法及第三法小杯法。
1.5凡检查溶出度的制剂,不再进行崩解时限的检查。
2.仪器与用具2.1溶出度仪2.1.1仪器的组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详见中国药典2000年版二部附录X C。
2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。
2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。
2.1.3.1溶出度校正片分崩解型和非崩解型两种,崩解型为泼尼松片,非崩解型为水杨酸片。
目前国内仅有非崩解型校正片。
2.1.3.2校正前,应先调式所用仪器。
2.1.3.3溶剂:磷酸盐缓冲液(PH7.4)。
配制方法见中国药典2000年版二部附录XV D,要求PH值为7.40±0.05,临用前脱气。
2.1.3.4对照品溶液的制备取溶出度校正用水杨酸片1片,精密称定,置乳体中,研细,精密称取适量(约相当于水杨酸10mg),置100ml量瓶中,加乙醇1ml,摇匀,加溶剂适量,经超声处理30分钟,使水杨酸溶解,加溶剂到刻度,摇匀,经滤纸(不宜使用滤膜)滤过,取续滤液为对照品溶液。
《中国药典》二部凡例

二、国家药品标准由凡例与正文及其引用 的附录共同构成。本部药典收载的凡例、 附录对药典以外的其他化学药品国家标准 具有同等效力。 三、凡例是为正确使用《中国药典》进行 药品质量检定的基本原则,是对《中国药 典》正文、附录及质量检定有关的共同问 题的统一规定。
四、凡例附录中采用“除另有规定外”这 一用语,表示存在与凡例或附录有关规定 不一致的情况时,则在正文中另作规定, 并按此规定执行。表示存在与凡例或附录有关规定
《中国药典》凡 例
《中华人民共和国药典》(以下简 称《中国药典》)是国家监督管理药 品质量的法定技术标准。是我国药品 标准体系的核心,是药品研究、生产、 经营、使用和监管的法定依据。
“凡例”是解释和使用《中国药典》正 确进行质量检定的基本指导原则,并把与 正文、附录及质量检定有关的共性问题加 以规定,避免在全书中重复说明。“凡例” 中的有关规定具有法定的约束力。
对于生产过程中引入的有机溶剂,应在后 续的生产环节予以有效去除。除正文已明 确列有“残留溶剂”检查的品种必须依法 进行项检查外,其他未在“残留溶剂”项 下明确列出的有机溶剂与未在正文中列有 此项检查的各品种,如生产过程中引入或 产品中残留有机溶剂,均应按附录“残留 溶剂测定法”检查并应符合相应溶剂的限 度规定。 供直接分装成注射用无菌粉针的原料药, 应按照注射剂项下相应的要求进行检查, 并应符合规定。 各类制剂,除别有规定外,均应符合各制 剂项下通则项下有关的各项规定。
正
文
八、正文系根据药物自身的理化与生物学特性, 按照批准的处方来源、生产工艺、贮藏运输等条 件所制定的,用以检测药品质量是否达到用药要 求并衡量其质量是否稳定均一的技术规定。 九、正文项下根据品种和剂型的不同,按顺序可 分别列有:(1)品名(包括中文名、汉语拼音与 英文名);⑵有机药物的结构式;⑶分子式与分 子量;⑷来源或有机药物的化学名称;⑸含量或 效价规定;(6)处方;(7)制法;(8)性状; (9)鉴别;(10)检查;(11)含量或效价测 定;(12)类别;(13)规格;(14)贮藏; (15)制剂等。
SOP-06-QC-032-02残留溶剂测定法操作规程(EP、BP)

目录1.目的:--------------------------------------------------------------------22.范围:--------------------------------------------------------------------23.职责:--------------------------------------------------------------------24.内容:--------------------------------------------------------------------2 4.1简述:-------------------------------------------------------------------2 4.2仪器与用具:-------------------------------------------------------------2 4.3试剂:-------------------------------------------------------------------3 4.4操作:-------------------------------------------------------------------34.5结果与判定:-------------------------------------------------------------65.相关程序:---------------------------------------------------------------106.相关记录:---------------------------------------------------------------107.参考资料:---------------------------------------------------------------108.附件:-------------------------------------------------------------------109.历史和修订记载:---------------------------------------------------------201.目的:建立残留溶剂测定法操作规程(EP、BP),使测定过程按规定程序有序进行,确保检验结果的准确性和可靠性。
原料药残留溶剂试验的要求及常见问题分析-2011.4-北京周立春.pdf

举例:头孢泊肟酯
残留溶剂 照残留溶剂测定法(附录Ⅷ P)测定。 甲醇、乙腈、丙酮、二氯甲烷、异丙醇、丁酮、乙酸乙 酯、四氢呋喃、乙酸丁酯、1,2-二氯乙烷、乙酸异丙酯、 苯、四氯化碳、环己烷、二氧六环、甲基异丁基酮、吡 啶、甲苯 色谱条件与系统适用性试验 略 内标溶液的制备 取正丙醇适量,用二甲基亚砜稀释制成 每1ml中约含200μg的溶液,作为内标溶液。
顶空进样:
原理:将含有挥发性组分的样品 置于密闭系统中,在一定温 度下使样品中的挥发性组分 在气-液或气-固两相甚至气液-固三相中的分配达到平衡, 然后取凝聚相上端的气体送 入气相色谱仪进行分析 优点:干净。样品中不挥发组分 不影响GC分析。减轻污染。 要求:样品至少在顶空条件下溶 解。
K
1 p i
0 i
p0i为纯溶质的蒸气 压 γi为组分的活度系 数
顶空条件的选择
顶空温度:残留溶剂的沸点较高,顶空温度也应 相应提高;但应兼顾供试品的热分解特性,尽量 避免供试品产生的挥发性热分解产物对测定的干 扰。 顶空平衡时间:一般为30~45分钟,以保证供试 品溶液的气-液两相有足够的时间达到平衡。顶 空时间通常不宜过长,如超过60分钟,可能引起 顶空瓶的气密性变差,导致定量准确性的降低。 传输管温度:对于有传输管的顶空进样器,传输 管温度应适当,通常设定在110℃~120℃。 对照品溶液与供试品溶液必须使用相同的顶空条 件。
原料药残留溶剂试 验的要求及常见问 题分析
周立春 2011.4
主要内容
1. 2. 3. 4.
前言 2010版药典中残留溶剂的相关内容 残留溶剂的方法学研究 残留溶剂测定的常见问题
前 言
1) 2) 3)
残留溶剂定义 残留溶剂的分类 药物中残留溶剂的特点
30环氧乙烷残留检验操作规程

30环氧乙烷残留检验操作规程一、检验目的本操作规程旨在规范环氧乙烷残留检验的操作步骤,确保环氧乙烷产品符合相关标准和要求。
二、适用范围本操作规程适用于环氧乙烷的生产和质检过程中对其残留量进行检验。
三、设备和试剂1.环氧乙烷残留检验仪器:气相色谱仪、样品瓶、移液器等。
2.试剂:二氯甲烷、丙酮、乙醇。
四、操作步骤1.样品制备(1)取适量环氧乙烷样品,将其转移到样品瓶中。
(2)将样品瓶密封并进行标记,确保样品不会泄漏或混入其他物质。
2.样品提取(1)在实验室条件下,使用合适的提取方法将环氧乙烷样品中的残留物提取出来。
可以使用溶剂提取法或固相萃取法等。
(2)将提取的残留物转移到样品瓶中,确保样品不会泄漏或混入其他物质。
3.样品预处理(1)将样品瓶中的残留物与二氯甲烷进行适量的稀释。
稀释倍数视实际样品情况而定,一般为1:10或1:100。
(2)使用适量的丙酮进行洗涤,以去除残留物中的杂质。
(3)使用适量的乙醇进行再次洗涤,以去除残留物中的残留丙酮。
(4)将预处理后的样品置于离心管中进行离心分离。
4.气相色谱检测(1)将分离后的样品取出,使用移液器等装置将其转移到气相色谱仪中。
(2)设置气相色谱仪的相关参数,如进样模式、流速、温度等,确保检测精度和准确性。
(3)进行样品的气相色谱检测,并记录检测结果。
五、质量控制1.每批次检验前,应进行仪器的校验和调试,确保检测结果准确可靠。
2.每个样品的提取和处理过程应进行严格控制,避免样品污染和误差。
3.定期参与相关质量控制方案,与其他实验室比对检测结果,确保测试结果的一致性和可靠性。
六、数据处理和结果评定1.将检测到的环氧乙烷残留浓度与相关标准比较,判断样品是否符合要求。
2.统计和记录检验结果,并进行结果分析和评价。
3.根据结果评价,及时采取相应的措施进行调整和改进。
七、安全注意事项1.在操作过程中,严格遵守实验室安全操作规程,保持良好的实验操作习惯。
2.使用有害溶剂时,应戴好防护手套、护目镜等个人防护装备,避免溶剂对身体的伤害。
溶剂残留检测操作规程
本规程适用于气相色谱-FID检测1 标准曲线的绘制1.1 样品的制备a 用移液管准确移取乙酸乙酯、异丙醇、甲苯、二甲苯各2ml到25ml标准瓶中;b 将标准瓶密封后摇匀,用微量注射器准确抽取0.4μl混合液分别注入25ml标准瓶中后密封好;c 将上述标准瓶放在80±5℃的烘箱中烘烤30min以上使其完全气化;d 在进样前2-5min将标准瓶从烘箱中取出待用。
1.2 样品的采集按气相色谱的操作规程进行操作,分析条件如下:a 采用FID检测器以及毛细管柱进行分析;b 载气为氮气,气源总压力为0.7Mpa,流量控制表压为50Kpa;c 柱温初始温度为36℃,采用程序升温。
升温程序为:初始温度为36℃,保温4min后以30℃/min的速度升温至90℃,保温1min。
;d 进样口温度INJ为200℃、检测器温度DET为250℃;e 空气、氢气气源总压为0.2Mpa,空气的流量控制表压为50Kpa、氢气为60Kpa;f 进样量分别为0.3ml、0.4ml、0.5ml、0.6ml、0.7ml气体。
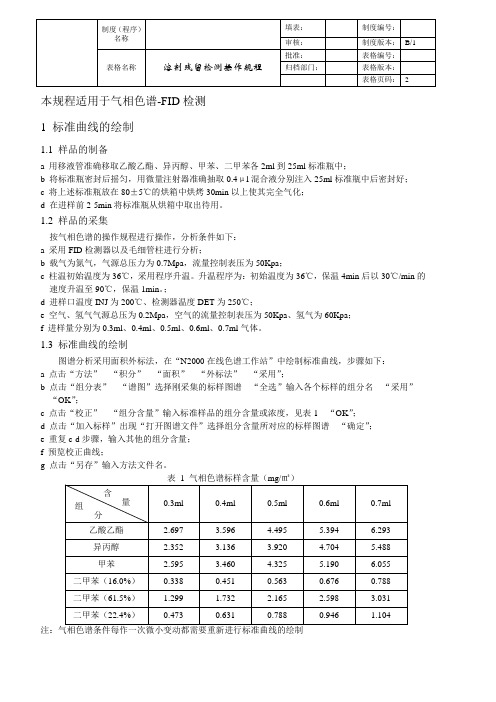
1.3 标准曲线的绘制图谱分析采用面积外标法,在“N2000在线色谱工作站”中绘制标准曲线,步骤如下:a 点击“方法”---“积分”---“面积”---“外标法”---“采用”;b 点击“组分表”---“谱图”选择刚采集的标样图谱---“全选”输入各个标样的组分名---“采用”---“OK”;c 点击“校正”---“组分含量”输入标准样品的组分含量或浓度,见表1---“OK”;d 点击“加入标样”出现“打开图谱文件”选择组分含量所对应的标样图谱---“确定”;e 重复c-d步骤,输入其他的组分含量;f 预览校正曲线;g 点击“另存”输入方法文件名。
注:气相色谱条件每作一次微小变动都需要重新进行标准曲线的绘制2 待测样的溶剂残留测定2.1 待测样的制备a 取待测样膜10cm×10cm,剪碎后装入25ml的标准瓶中密封;b 将标准瓶放在80±5℃的烘箱中烘烤30min以上使其残留溶剂完全气化;c在进样前2-5min将标准瓶从烘箱中取出冷却待用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1. 目的:建立残留溶剂测定法(二部)检验标准操作规程,并按规程进行检验,保证检验操作规范化。
2. 依据:2.1. 《中华人民共和国药典》2010年版二部。
3.范围:适用于所有用残留溶剂测定法(二部)测定的供试品。
4. 责任:检验员、质量控制科主任、质量管理部经理对本规程负责。
5.正文:5.1. 药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。
药品中常见的残留溶剂及限度见附表1,除另有规定外,第一、第二、第三类溶剂的残留限度应符合表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范、药品生产质量管理规范(GMP)或其他基本的质量要求。
5.2. 本法照气相色谱法(附录Ⅴ E)测定。
5.3. 色谱柱5.3.1. 毛细管柱:除另有规定外,极性相近的同类色谱柱之间可以互换使用。
5.3.1.1. 非极性色谱柱:固定液为100%的二甲基聚硅氧烷的毛细管柱。
5.3.1.2. 极性色谱柱:固定液为聚乙二醇(PEG-20M)的毛细管柱。
5.3.1.3. 中性色谱柱:固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。
5.3.1.4. 弱极性色谱:柱固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷的毛细管柱等。
5.3.2. 填充柱:以直径为0.18~0.25mm 的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。
5.4. 系统适用性试验。
5.4.1. 用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000;填充柱的理论板数一般不低于1000。
5.4.2. 色谱图中,待测物色谱峰与其相邻色谱峰的分离度应大于1.5。
5.4.3. 以内标法测定时,对照品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)不大于5%;若以外标法测定,所得待测物峰面积的RSD应不大于10%。
5.5. 供试品溶液的制备。
5.5.1. 顶空进样:除另有规定外,精密称取供试品0.1~1g,通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;根据供试品和待测溶剂的溶解度,选择适宜的溶剂且应不干扰待测溶液的测定。
根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。
5.5.2. 溶液直接进样:精密称取供试品适量,用水或合适的有机溶剂使溶解;根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。
5.6. 对照品溶液的制备:精密称取各品种项下规定检查的有机溶剂适量,采用与制备供试品溶液相同的方法和溶剂制备对照品溶液;如用水作溶剂,应先将待测有机溶剂溶解在50%二甲基亚砜或N,N-二甲基甲酰胺溶液中,再用水逐步稀释。
若为限度检查,根据残留溶剂的限度规定确定对照品溶液的浓度;若为定量测定,为保证定量结果的正确性,应根据供试品中残留溶剂的实际残留量确定对照品溶液的浓度;通常对照品溶液的色谱峰面积不宜超过供试品溶液中对应的残留溶剂的色谱峰面积2倍。
必要时,应重新调整供试品溶液或对照品溶液的浓度。
5.7. 测定法。
5.7.1. 第一法(毛细管柱顶空进样等温法)。
5.7.1.1. 当需要检查有机溶剂的数量不多,且极性差异较小时,可采用此法。
5.7.1.2. 色谱条件:柱温一般为40~100℃;常以氮气为载气,流速为每分钟1.0~2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;如采用火焰离子化检测器(FID),温度为250℃。
5.7.1.3. 测定法:取对照品溶液和供试品溶液,分别连续进样不少于2次,测定待测峰的峰面积。
5.7.1.4. 对色谱图中未知有机溶剂的鉴别,可参与附表2进行初筛。
5.7.2. 第二法(毛细管柱顶空进样系统程序升温法)。
5.7.2.1. 当需要检查的有机溶剂数量较多,且极性差异较大时,可采用此法。
5.7.2.2. 色谱条件:柱温一般先在40℃维持8分钟,再以每分钟8℃的升温速率升至120℃,维持10分钟;以氮气为载气,流速为每分钟2.0ml;以水为溶剂时顶空瓶平衡温度为70~85℃,顶空瓶平衡时间为30~60分钟;进样口温度为200℃;如采用FID检测器,进样口温度为250℃。
5.7.2.3. 具体到某个品种的残留溶剂检查时,可根据该品种项下残留溶剂的组成调整升温程序。
5.7.2.4. 测定法:取对照品溶液和供试品溶液,分别连续进样不少于2次,测定待测峰的峰面积。
5.7.2.5. 对色谱图中未知有机溶剂的鉴别,可参考附表3进行初筛。
5.7.3. 第三法(溶液直接进样法)。
5.7.3.1. 可采用填充柱,亦可采用适宜极性的毛细管柱。
5.7.3.2. 测定法:取对照品溶液和供试品溶液,分别连续进样2~3次,测定待测峰的峰面积。
5.7.3.3. 计算法。
5.7.3.3.1. 限度检查:除另有规定外,按品种项下规定的供试品溶液浓度测定。
以内标法测定时,供试品溶液所得被测溶剂峰面积与内标峰面积之比不得大于对照品溶液的相应比值。
以外标法测定时,供试品溶液所得被测溶剂峰面积不得大于对照品溶液的相应峰面积。
5.7.3.3.2. 定量测定:按内标法或外标法计算各残留溶剂的量。
5.8. 【附注】。
5.8.1. 除另有规定外,顶空条件的选择。
5.8.1.1. 应根据供试品中残留溶剂的沸点选择顶空平衡温度。
对沸点较高的残留溶剂,通常选择较高的平衡温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物对测定的干扰。
5.8.1.2. 顶空平衡时间一般为30~45分钟,以保证供试品溶液的气-液两相有足够的时间达到平衡。
顶空平衡时间通常不宜过长,如超过60分钟,可能引起顶空瓶的气密性变差,导致定量准确性的降低。
5.8.1.3. 对照品溶液与供试品溶液必须使用相同的顶空条件。
5.8.2. 定量方法的验证。
5.8.2.1. 当采用顶空进样时,供试品与对照品处于不完全相同的基质中,故应考虑气液平衡过程中的基质效应(供试品溶液与对照品溶液组成差异对顶空气-液平衡的影响)。
由于标准加入法可以消除供试品溶液基质与对照品溶液基质不同所致的基质效应的影响,故通常采用标准加入法验证定量方法的准确性;当标准加入法与其他定量方法的结果不一致时,应以标准加入法的结果为准。
5.8.3. 干扰峰的排除:供试品中的未知杂质或其挥发性热降解物易对残留溶剂的测定产生干扰。
干扰作用包括在测定的色谱系统中未知杂质或其挥发性热降解产物与待测物的结构相同(如甲氧基热裂解产生甲醇)。
当测定的残留溶剂超出限度,但未能确定供试品中是否有未知杂质或其挥发性热降解物对测定有干扰作用时,应通过试验排除干扰作用的存在。
对第一类干扰作用,通常采用在另一种极性不同的色谱柱系统中对相同供试品再进行测定,比较不同色谱系统中对相同供试品再进行测定,比较不同色谱系统中测定结果的方法。
如两者结果一致,则可以排除测定中有共出峰的干扰。
对第二类干扰作用,通常要通过测定已知不含该溶剂的对照样品来加以判断。
5.8.4. 含氮碱性化合物的测定。
5.8.4.1. 普通气相色谱仪中的不锈钢管路、进样器的衬管等对有机胺等含氮碱性化合物具有较强的吸附作用,致使其检出灵敏度降低,应采用惰性的硅钢材料或镍钢材料管路;采用溶液直接进样法测定时,供试品溶液应不呈酸性,以免待测物与酸反应后不易汽化。
5.8.4.2. 通常采用弱极性的色谱柱或其填料预先经碱处理过的色谱柱分析含氮碱性化合物,如果采用胺分析专用柱进行分析,效果更好。
5.8.4.3. 对不宜采用气相色谱法测定含氮碱性化合物,如N-甲基吡咯烷酮等,可采用其他方法如离子色谱法等测定。
5.8.5. 检测器的选择:对含卤素元素的残留溶剂如三氯甲烷等,采用电子捕获检测器(ECD ),易得到高的灵敏度。
5.8.6. 由于不同的实验室在测定同一供试品时可能采用了不同的实验方法,当测定结果处于合格与不合格边缘时,以采用内标或标准加入法为准。
5.8.7. 顶空平衡温度一般应低于溶解供试品所用溶剂的沸点10℃以下,能满足检测灵敏度即可;对于沸点过高的溶剂,如甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮等,用顶空进样测定的灵敏度不如直接进样,一般不宜用顶空进样方法测定。
5.8.8. 利用保留值定性是气相色谱中最常用的定性方法。
色谱系统中载气的流速、载气的温度和柱温等的变化都会使保留值改变,从而影响定性结果。
校正相对保留时间(RART )只受柱温和固定相性质的影响,以此作为定性分析参数较可靠。
应用中通常选用甲烷测定色谱系统的死体积(0t )RART='t t t t R R -- 式中 R t 为组分的保留时间; R t '为参比物的保留时间。
附表1:药品中常见的残留溶剂及限度。
①通常含有60%间二甲苯、14%对二甲苯、9%邻二甲苯和17%乙苯。
②药品生产企业在使用时应提供该类溶剂在制剂中残留水平的合理性论证报告。
附表2 常见有机溶剂在等温法测定时相对于丁酮的保留值参考值附表3 常见有机溶剂在程序升温法测定时相对于丁酮的保留值参考值注:附表2、3中数据未非极性的SPB-1柱(30m×0.32mm,1.0μm)和极性的HP-INNOWAX柱(30m×0.32mm,0.5μm)测定的结果。