基于RAD测序的枸杞SNP分布特征分析
基于GBS测序开发SNP在植物上的应用进展

基于GBS测序开发SNP在植物上的应用进展作者:薛晓杰杜晓云盖艺唐岩孙燕霞宋来庆姜中武来源:《江苏农业科学》2020年第13期摘要:基因分型测序(genotyping by sequencing,GBS)因具有实现相对简单、成本较低、可产生高通量SNP的优点而受到青睐。
单核苷酸多态性(single nucleotide polymorphism,SNP)因简单、快速、特异性强、稳定遗传、便于检测等特点已成为目前最广泛使用的分子标记之一。
本文就利用GBS技术开发SNP分子标记近年来在亲缘关系评价、重要农艺性状鉴定、遗传多样性研究、遗传图谱构建以及基因定位等方面在国内外的研究进展进行综述,并对其今后的研究提出展望,以期为其在植物上的更广泛应用提供参考。
关键词:SNP分子标记;GBS技术;简化基因组测序;遗传图谱构建;QTL定位;基因定位中图分类号:S184 文献标志码: A文章编号:1002-1302(2020)13-0062-07收稿日期:2020-040-02基金项目:山东省外专双百计划(编号:WST2018013);山东省重点研发项目(编号:2018GHZ005)。
作者简介:薛晓杰(1994—),女,山东烟台人,硕士研究生,主要从事果树分子与生理技术研究,E-mail:xuexiaojieYT@;共同第一作者:杜晓云(1979—),女,山西吕梁人,博士,高级农艺师,主要从事果树生物技术育种研究,E-mail:duxiaoyunduzi@。
通信作者:姜中武,博士,研究员,主要从事果树育种与栽培技术研究。
E-mail:jiangzhongwu@。
随着分子生物学的发展,分子标记技术发展迅速,基因组测序技术日渐成为标记开发的重要手段。
简化基因组测序(GBS)技术近年发展起来。
GBS技术可以捕获基因组重要区域,获得大量单核苷酸多态性(single nucleotide polymorphism,SNP),且无需已知基因组信息[1]。
基于ITS条形码序列对枸杞杂交种的早期鉴定

基于ITS条形码序列对枸杞杂交种的早期鉴定石志刚;万如;李彦龙;王亚军【摘要】采用改进CTAB法提取枸杞(Lycium Linn)叶片DNA,利用合成的特异引物对其DNA中nrDNAITS区进行扩增、克隆,对目的片段进行测序分析,以期利用ITS条形码序列对枸杞杂交种进行早期鉴定.结果表明,7份枸杞属不同种间杂交种的ITS序列长度变异范围为558~632 bp,平均为610 bp.排序后的总长度为632 bp,整个转录间隔区(ITS1 +ITS2)对位排列后总长度为478 bp,共有138个变异位点,ITS1和ITS2分别为85和53个,占28.9%;保守位点340个,占71.1%;有6个信息位点,占1.3%;51个转换位点,12个颠换位点,其中ITS1区的信息位点所占比例低于ITS2区,而ITS1区的转换位点与颠换位点比值高于ITS2区.通过对宁夏枸杞、北方枸杞、黑果枸杞及其种间杂交育种产生的杂交后代分析,得出基于ITS聚类分析能够初步判别杂交后代与父母本的亲缘关系与差异,可以作为早期鉴定枸杞属不同种间杂交种后代的方法之一.【期刊名称】《湖北农业科学》【年(卷),期】2016(000)019【总页数】3页(P5075-5077)【关键词】枸杞(Lycium Linn);ITS序列;鉴定;杂交育种【作者】石志刚;万如;李彦龙;王亚军【作者单位】宁夏农林科学院,银川 750002;宁夏农林科学院,银川 750002;宁夏农林科学院,银川 750002;宁夏农林科学院,银川 750002【正文语种】中文【中图分类】S567.1+9枸杞(Lycium Linn)富含枸杞多糖、甜菜碱、类胡萝卜素、多种不饱和脂肪酸等多种有效成分,具有抗氧化、抗肿瘤、延缓衰老、增强免疫力、软化血管、降低血脂等功效,是中国重要的药食同源功能型特色植物资源[1]。
宁夏是枸杞的原产地和主产区,截至2015年底,宁夏种植规模达到5.71万hm2,占全国种植总面积的42.5%;枸杞干果产量13万t,占全国总产量的48%;年出口量6 500 t,出口额达7 000万美元,分别占全国出口量和出口额的65%和58%;枸杞生产总值突破50亿元。
基于ITS条形码序列的枸杞属植物鉴定

基于ITS条形码序列的枸杞属植物鉴定摘要:采用改进CTAB法提取枸杞(Lycium Chinense Mill.)叶片DNA,利用合成的特异引物对其DNA中nrDNA ITS区进行扩增,利用ITS条形码序列,对枸杞属种质资源进行鉴定,分析其亲缘关系。
结果表明,测序得到了17份枸杞属近缘种的ITS条形码序列,整个ITS序列长度变异范围为603~632 bp,平均为624 bp,整个转录间隔区(ITS1+ITS2)对位排列后总长度为480 bp,有194个变异位点,占40%;保守位点288个,占60%。
聚类分析结果表明,17份种质资源可分为5个大类群。
基于ITS条形码序列分析在鉴定枸杞属种质遗传多样性及其亲缘关系具有一定的优越性。
关键词:枸杞属(Lycium L.);ITS序列;DNA测序;鉴定中图分类号:S567.1+9 文献标识码:A 文章编号:0439-8114(2016)22-5966-03DOI:10.14088/ki.issn0439-8114.2016.22.059Identification of Lycium L. Germplasm Resources Based on nrDNA ITS SequenceSHI Zhi-gang,WAN Ru,LI Yan-long,WANG Ya-jun(Ningxia Academy of Agriculture and Forestry Sciences,Yinchuan 750002,China)Abstract:Adopting improved CTAB to acquire DNA of leaf of Lycium Chinense Mill.,and making use of composite special primers to propagate and clone nrDNA ITS area of DNA,and then sequencing analysis of the target segment. To identify genetic diversity of Lycium L. germplasm resources by the nrDNA ITS sequence. The results showed that the nrDNA ITS sequence of 17 Lycium L. was gained for the first time;the variation length of the whole ITS sequence was 603 to 632 bp,and the average was 624 bp;the total length of whole transcribe partition area(ITS1 and ITS2)sequence alignmentwas 480 bp,and there were 194 variable sites accounting for 40.4%,and 286 conserved sitesaccounting for 59.6%. Based on the nrDNA ITS sequence analysis of 17 Lycium L. germplasm resources,it was obvious that they could be divided into 5 groups. The nrDNA ITS sequence analysis is a advantageous way to study genetic diversity among Lycium L. germplasm resources.Key words:Lycium L.;Internal transcribed spacer;measure the sequence of DNA;identification枸杞(Lycium Chinense Mill.)?儆谇芽疲?Solanaceae)族(Solaneae Reichb.)枸杞属(Lycium L.),枸杞属植物在全球的分布约有80种,多分布于南、北美洲,以美国亚利桑那州和阿根廷形成两个分布中心;欧亚大陆约10余种。
基于近红外光谱的中宁枸杞子判别分析

could be clustered into
groups using the Mahalanobis distances in combination with the Ward’S method:one group consisting of those from
Zhongning,Ningxia
文章编号:1002-6630(2014)02—0164-00
枸杞(L脚an barbarian L)为茄科构杞属多分枝灌木(蚴,
枸杞子为枸杞的干燥成熟果实口】,具有味甘、性平,滋
高。建立自动、准确、环保的枸杞子产地鉴别方法,以 保证宁夏中宁枸杞子的品质,具有十分重要意义。近红 外光谱对应分子基频振动的倍频和组合频,其特征随 着样品成分含量的变化而变化n 41,具有无损、绿色、实 时监控的特点n熨,非常适合农产品和食品的快速判别分 析【l 61。目前有关枸杞子近红外光谱产地鉴定报道较少, 汤丽华等¨∞采用近红外光谱技术结合简易分类(simple
其他地区的平均值高出0.585、0.378、0.0197、0.019 mg/g。
峋2音∑(‰一暑)(xjk一弓)。xr、■分别为第f个和箭
个样本的行向量,x。。为第i个样本的第k个特征变量,x陆 为第,个样本的第k个特征变量,工。、x分别为第f个和第 ,个样本所有特征变量的均值。
1.5
聚类分析
聚类分析具有无管理模式识别方法的特点,利用相
万方数据
166
2014,V01.35,No.02
食品科学
键
血 H g № 理 蜒 卅 导 耐 * 祟
※分析检测
不同地区枸杞子化学成分的差异,可能是由于其生长的 生态环境不同而引起的。
2.1.2
光谱预处理对判别分析结果的影响
寻找SNP——RAD tags及其应用

质谱 法
等位基 因特异
PCR
检测 SNP
实时荧 光PCR
RFLP
传统SNP检测技术的不足
传统的SNP检测技术低通量、低效率,并且或 多或少的都需要已知的基因组背景信息,使得难 以从基因组背景信息不充分的非模式生物中发现
SNPs并进行高效和简洁的基因分型。
Contents
第二代测序技术
以高第通一代量为特征的第二代测序技术又称“下一代”测序 技术测(序“Next-generation” sequencing technology),能
Rainbow trout
westslope cutthroat trout
RAD tags与渐渗杂交
1992年,Thomas等采用mtDNA限制性内切酶分析,发现 Apacbe throut与Rainbow trout 的关系比westslope cutthroat trout 更近。并用等位酶标记分析发现Apacbe throut与Rainbow trout 在四个种群中有渐渗杂交。
RAD tags与系统地理学
采用COI构树
使用来自20个W. smithii 种群和两 个外群 Wyeomyia mitchellii 和 Wyeomyia vanduzeei的1176bp的 COI片段,采用最大简约法、最大 似然法和贝叶斯法来推断这些种 群间的系统发生关系。节点处的 数字显示了该节点获三种方法各 自的支持率。
一、基因组DNA的提取
Etter等人建议采用Qiagen公司的DNeasy Blood & Tissue Kit试剂盒或相似的产品来提取和纯化DNA,以产生纯净、高分 子量和无RNA的DNA样品。确保采用RNAase处理样品以去除 残留的RNA。最佳的DNA浓度是25ng/μl或更高。
基于GBS测序的全基因组SNP揭示贵州地方茶组植物资源的亲缘关系
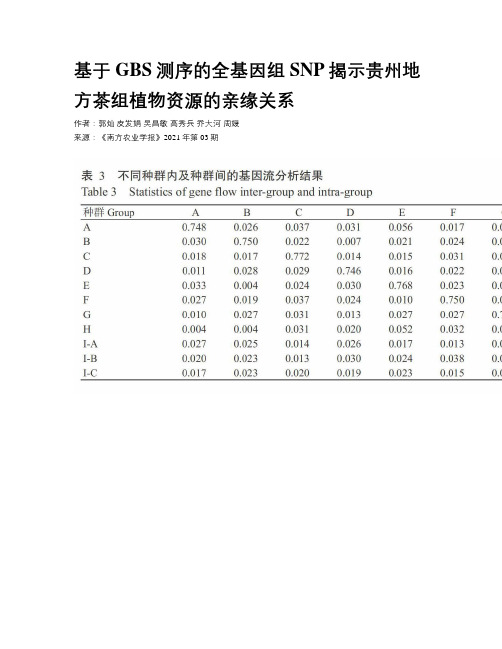
基于GBS测序的全基因组SNP揭示贵州地方茶组植物资源的亲缘关系作者:郭灿皮发娟吴昌敏高秀兵乔大河周媛来源:《南方农业学报》2021年第03期摘要:【目的】分析贵州地方茶组植物资源的亲缘关系,为明确贵州地方茶组植物资源的遗传关系及其保护、利用提供科学依据。
【方法】以从贵州省内不同区域收集的41份茶组植物资源及贵州省茶叶研究所茶树种质资源圃保存的18份省内外育成茶树品种为材料,利用基于GBS(Genotyping by sequencing)的简化基因组测序技术对其基因组SNP位点进行检测,基于获得的高质量SNP位点对这些材料进行遗传特征分析。
【结果】从59份茶组植物材料获得45.84 Gb高质量序列(Clean reads)数据,平均每个材料为795.6 Mb,约占改良版茶树基因组大小(2.93 Gb)的26.5%,平均比对率为72.62%,经过滤后得到248772个高质量SNP位点,其中83.98%的高质量SNP位点分布在基因间区,16.02%分布于基因区;有22614个SNP 位点分布在内含子,15038个SNP位点分布在外显子区,2203个SNP位点分布在非翻译区(UTR)。
59份茶组植物材料的观察杂合度(Ho)为0.016~0.081,期望杂合度(He)为0.006~0.064,F为-0.331~0.737。
主成分分析结果、系统发育进化树构建情况及遗传结构分析结果均显示59份茶组植物材料可分为3个类群,其中全部茶种(Camellia sinensis)材料归在一个类群、疏齿茶(C. remotiserrata)和大厂茶(C. tachangensis)归在一个类群、9份突肋茶(C. costata)单独归在一个类群,但疏齿茶与大厂茶及两个区域的大厂茶均处于独立的亚类群,此外茶种中的阿萨姆变种(C. sinensis var. assamica,CSA)和中国变种(C. sinensis var. sinensis,CSS)也处于不同的进化分支;突肋茶与疏齿茶和大厂茶的亲缘关系较其与茶种的亲缘关系更近。
宁夏主栽枸杞根际土壤微生物群落特征分析

宁夏主栽枸杞根际土壤微生物群落特征分析宁夏主栽枸杞(Lycium barbarum L.)根际土壤微生物群落特征分析引言:枸杞是宁夏农业的重要作物之一,因其丰富的营养和药用价值而备受关注。
土壤是植物生长的基础,而土壤中的微生物群落对于植物的生长发育和健康状态有着重要影响。
因此,研究枸杞根际土壤微生物群落特征对于了解枸杞生长环境和提高产量质量具有重要意义。
方法:本研究选择了宁夏某枸杞种植基地的根际土壤样品作为研究对象,采用高通量测序技术对其进行了微生物群落特征分析。
首先,从不同采样点采集了若干土壤样品,并进行了DNA提取。
随后,利用16S rRNA基因和ITS基因作为引物,通过PCR扩增获得微生物DNA条形码。
最后,经过Illumina MiSeq平台测序后,对测序结果进行分析。
结果:通过测序和分析,我们获得了宁夏根际土壤微生物群落的丰富度、多样性和组成情况。
研究发现,根际土壤中的微生物多样性非常丰富,包括细菌、真菌和古菌等。
其中,细菌是最主要的群落成员,占总微生物数量的大部分。
真菌的种类相对较少,但在土壤养分循环和植物生长中具有重要作用。
此外,我们还分析了不同样品之间的微生物群落差异。
结果显示,不同样品之间存在一定的差异,主要表现为微生物群落组成的不同。
这可能是由于土壤中的环境因素(如土壤pH 值、含水量等)和枸杞根系分泌的物质影响微生物群落的结构。
讨论:本研究结果表明,宁夏主栽枸杞的根际土壤微生物群落具有较高的多样性和丰度。
在这个群落中,细菌是最主要的成员,但真菌也对于土壤养分循环具有重要作用。
此外,不同样品之间的微生物群落差异可能受到环境因素和植物根系分泌物质的影响。
这些结果对于枸杞的种植和土壤管理具有一定的指导意义。
首先,保持土壤微生物群落的多样性和丰度是保证植物健康生长的关键。
其次,合理调节土壤环境因子,如pH值和水分含量等,有助于维持良好的微生物群落结构。
此外,注意植物的根际分泌物质的影响,可以更好地优化土壤环境,提高枸杞产量和质量。
基于近红外光谱的枸杞化学成分定量分析

1.3.3 近红外光谱的定量分析方法
叠情况严重,样品因种间差异和种植环境因素等的不
本文采用偏最小二乘回归法(PLS)作为枸杞近 同,主要化合物的含量也会有差别。因此,必须对原
红外光谱的定量分析方法。偏最小二乘回归(PLS)[7~8] 始光谱进行处理,才能利用近红外光谱贵枸杞化学成
是近红外光谱分析中应用最多的一种建模回归方法, 分进行定量分析。本文采用一阶导数+五点平滑+矢量
蛋白质:GB 5009.5-2010;脂肪:GB 5009.6-2003;总 如图 1 所示。
糖:GB/T 5009.7-2008;还原糖:GB/T 5009.7-2008;
由图 1 可以看出,光谱图中由上至下08;维生素 C:2, 4-二硝基苯肼 北、中宁、惠农、内蒙、甘肃、青海、同心、南梁 8
1.4 衡量校正模型的参数
对物质成分做定量分析的研究还是较少的,主要集中
本试验对枸杞样品采用全波段光谱扫描,建立校
于药品、粮食和茶叶等方面,对于枸杞成分的近红外 正模型后,必须通过对验证集样品的测量来判断校正
定量分析还未见有报道,因此本试验利用近红外光谱 模型的稳定性和准确性,校正模型验证常用的评估参
加突出,因此以该处理后的光谱信息作为定量分析的 成分变化范围,可以建立枸杞样品定量分析模型。
预测集。
表 1 枸杞样品化学成分的分析结果
Table 1 The chemical value of Lycium barbarum L
成分
样品数 最大值 最小值 平均值
N
M ax. M in. M ean
图 1 8 个产地枸杞的近红外光谱图 Fig.1 NIR spectra of Lycium barbarum Lfrom eight regions
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Hans Journal of Agricultural Sciences 农业科学, 2018, 8(7), 699-704Published Online July 2018 in Hans. /journal/hjashttps:///10.12677/hjas.2018.87105SNP Distribution Characteristic of ChineseWolfberry Based on RAD SequencingGuanghui Fan1,2, Hang Yu1, Zhanlin Wang1,21Qinghai Academy of Agriculture and Forestry, Qinghai University, Xining Qinghai2Qinghai Plateau Key Laboratory of Tree Genetics and Breeding, Xining QinghaiReceived: Jun. 20th, 2018; accepted: Jul. 3rd, 2018; published: Jul. 10th, 2018AbstractThe single-nucleotide polymorphisms (SNPs) in the genome of Lycium barbarum were identified using the high throughput sequencing technology based on the Illumina HiSeq2500 platform. A total of 5,780,671,000 bp high quality data were produced. All of the reads were assembled into 880,315 contigs with 295 bp average length. Using the contig assemblies as a reference, 721,813 SNPs were identified. Among the SNPs, transitions were 454,827, transversions were 266,986, and the value of Ts/Tv was 1.70. Among the SNPs, A/G (31.69%) was the most abundant, followed with C/T (31.32%), A/C (10.78%), G/T (10.75%), A/T (10.27%) and C/G (5.18%).KeywordsLycium, SNPs, RAD, High Throughput Sequencing基于RAD测序的枸杞SNP分布特征分析樊光辉1,2,虞杭1,王占林1,21青海大学农林科学院林业研究所,青海西宁2青海高原林木遗传育种实验室,青海西宁收稿日期:2018年6月20日;录用日期:2018年7月3日;发布日期:2018年7月10日摘要利用illumina hiseq2500平台,对枸杞进行了RAD测序并对其SNPs的数目和分布特征进行了分析和比较。
樊光辉等测序后共得到5,780,671,000 bp的高质量数据,经过组装后得到平均长度为295 bp的contig 880,315个。
采用软件进行SNP的检测后得到721,813个SNPs。
其中转换替换共有454,827个,占总数的63.01%,颠换共有266,986个,占总数的36.98%,转换和颠换的比例Ts/Tv为1.70。
所有的替换类型中A/G占的比例最高,为31.69%,其余类型所占的比例依次为C/T (31.32%), A/C (10.78%), G/T (10.75%), A/T(10.27%)和C/G (5.18%)。
关键词枸杞,SNPs,RAD标记,高通量测序Copyright © 2018 by authors and Hans Publishers Inc.This work is licensed under the Creative Commons Attribution International License (CC BY)./licenses/by/4.0/1. 引言枸杞为茄科枸杞属的灌木树种,生长在干旱和半干旱地区,盐碱生境和海岸带也有分布(Fukuda et al., 2001) [1]。
我国枸杞的分布地区基本集中在西北和其它干旱、半干旱地区以及一些盐碱地区(Jia et al., 2009)[2]。
枸杞对于恶劣的土壤和气候环境条件具有很强的适应性,能在极度干旱、含盐量高的土壤中正常生长,因此在北方,尤其是我国青海、内蒙、甘肃、宁夏和新疆地区作为于防沙、治沙和固沙的先锋树种(Zhao et al., 2004) [3]。
枸杞果实中含有很多活性物质,例如枸杞多糖,生物碱,黄酮和类胡萝卜素等物质,因而其具有促进免疫、抗衰老、抗肿瘤、清除自由基、抗疲劳、抗辐射、保肝、生殖功能保护和改善等多种作用(Inbaraj et al., 2010; Duan et al., 2010) [4] [5]。
枸杞果实和根很早就被用于治疗眼疾和炎症,也是治疗肝胆疾病和肾脏方面的疾病的传统药物(Hitchcock, 1932) [6]。
由于枸杞的生态和经济的双重作用,其在我国北方被广泛种植,甚至在宁夏,青海和新疆等地区已经成为地区经济收入的支撑产业。
SNP (Single-nucleotide polymorphisms),即单核苷酸多态性,是基因组序列中最丰富的DNA多态性,它们能够影响到蛋白的功能,因而也是很多疾病和表型特征变异的基础。
一般情况下,物种基因组内SNPs的分布和其分布特征是不均匀的,编码区的SNP分布频率要低于非编码区。
自然选择,遗传重组,突变率以及其他的因素都能影响到SNPs的分布密度(Nachman 2001) [7]。
SNPs中有一部分是能够通过影响蛋白的功能而影响到物种表型的变异,另一部分是对于表型没有任何影响的变异,这类变异称为沉默突变,或者同义突变。
这类突变数量巨大,并且具有稳定遗传的特点,因而在全基因组关联分析(GWAS, genome-wide association studies),遗传图谱的构建(Thomas, 2011) [8],QTL 分析(Garrett et al., 2012) [9],分子标记辅助育种(Thavamanikumar et al., 2011) [10]等反面都被广泛利用。
基于其在遗传学和基因组学方面的重要性,检测和研究SNP在基因组上的分布和特征也具有重要意义(Steele et al., 2008) [11]。
随着第二代通量测序技术的发展,其高通量、省时和高效的特点使得对SNP 的检测也步入新的阶段。
迄今为止,基于高通量测序的方法进行SNP的检测已经在很多物种中加以利用。
枸杞的植物化学,药理学和育种等方面研究已经比较深入,但其分子水平上的研究目前还处于起步阶段,其基因组SNPs方面的研究尚未见报道。
基础研究尤其是分子生物学方面的滞后在一定程度上影响了枸杞育种工作,所以其遗传学和基因组学方面的研究亟待进行。
基于枸杞在经济和生态方面的重要性,本文利用第二代高通量测序技术用RAD标记对枸杞的基因组进行了简化分析后查找了其基因组水平樊光辉等上的SNPs标记,这些标记可以用于下一步枸杞高密度遗传图谱,关联分析的标记开发和使用,为枸杞遗传学和基因组学研究奠定基础。
2. 材料和方法2.1. 植物材料和DNA提取宁夏枸杞(L. barbarum L.)新鲜叶子于2014年八月份采自青海诺木洪农场枸杞种质资源圃。
基因组DNA利用kitDP305 (天根,北京)提取。
提取后利用NanoDrop 2000 (Wilmington, DE, USA)和琼脂糖凝胶电泳进行质量检测。
2.2. 建库和测序用RAD (Restriction-site associated DNA-sequencing)测序方法将枸杞基因组进行简化(Baird, 2008) [12]。
取基因组DNA1ug,利用EcoRI内切酶进行消化(G|AATTC),然后加P1接头(可与EcoRI酶切DNA缺口互补);将连接有接头的所有片段混合后随即打断,电泳回收300 bp~700 bp的片段,然后末端平化后加A;加Solexa P2 Adapter,P2为局部双链分叉Y型DNA,可实现选择性的扩增同时含有P1和P2接头的RAD标记;PCR扩增两端分别含有P1和P2接头的tag序列。
制备好的测序库利用Qubit 2.0kit (Life Technologies, Carlsbad, CA, USA)检测质量,Agilent 2100 (AgilentTechnologies, Palo Alto, Calif)检测片段的大小。
检测后的测序库利用Illumina HiSeq2500 (Illumina Inc., San Diego, Calif)根据程序进行测序。
2.3. 数据质量控制和组装利用In-House scripts将低质量和重复的测序数据的去除,使用EcoRI (G|AATTC)酶切位点,对Clean reads进行去除重复处理后,统计去重后EcoRI捕获的Reads数。
利用Velvet Optimiser software (Zerbino D R and Birney E, 2008) [13],根据默认参数进行数据组装。
将带有酶切识别序列的reads进行聚类,并按照深度由大到小进行排序。
将深度高的reads作为种子进行聚类。
根据深度信息对聚类后的reads进行纠错、过滤重复区域等。
根据聚类的结果,将另一端的reads进行contig拼接,结合插入片段的大小和overlap 的关系,将拼接好的contig与另一端聚类的reads进行连接,组装成最终的contig序列。
2.4. SNP查找利用BWA软件(Li and Durbin 2009) [14]将测序的所有reads比对到组装好的序列上,比对结果经SAMTOOLS (Li et al., 2009) [15] [16]去除重复(参数:rmdup)。
Candidate sequence variation were filtered according to the following criteria:利用贝叶斯模型检测群体中的多态性位点,通过以下过滤和筛选得到高质量的SNPs:1) Q20质量控制(将质量值Q20即测序错误率大于1%的SNPs过滤掉);2) SNP的支持数(覆盖深度)在2~1000范围内;3) 缺失控制(将群体内SNP位点缺失率大于0.1的位点过滤掉)。