第七章化学键理论概述
化学中的化学键理论

化学中的化学键理论化学键是指原子间的吸引力力,是分子形成的基础。
化学键的形成、性质和断裂是化学反应的重要环节,也是化学研究的核心内容。
化学键理论是化学学科中的重要分支之一,它揭示了化学键的性质和本质,为化学科学的发展和应用提供了理论基础。
1. 传统化学键理论在传统的化学中,原子间的化学键是指开尔文的“亲和力”理论。
它将原子的吸引力定义为原子核和共享了某些电荷的电子间的作用力,是一个纯经验的观点。
它不是一个特别准确的预测性理论,但是仍然在一些情况下被广泛使用。
2. 共价键理论共价键理论是指两个原子通过共享电子共同发展出的化学键。
这一理论揭示了共价键的本质,即原子间电子的共享。
共价键通常用杂化轨道理论来解释。
杂化轨道理论认为,原子的价电子空壳轨道中的电子可能会混合成新的、更稳定的轨道,称为杂化轨道。
杂化轨道提供了一个更准确的方法来描述共价键——如在氨分子中,氮原子价电子空壳轨道和氢原子的原子轨道混合,产生了四个杂化轨道,分别用于和四个氢原子组成共价键。
3. 离子键理论离子键理论是指形成离子键的原理。
它是一种典型的原子或分子排斥的现象。
当两种化学物质中含有带电离子时,离子间会产生电吸引力,因此导致它们结合到一起,而这些带电离子被称为离子。
离子键通常发生在化合物中,如氯化钠(NaCl)和硫酸二钾(K2SO4)。
4. 金属键理论金属键是指金属中的化学键,通常是由金属离子通过共享电子形成金属键。
金属离子在结晶中排列成空间有序的三维结构,形成晶格。
这种排列方式为金属提供了良好的机械性能和导电性能,在大规模制造工业用金属和合金方面有着重要的应用。
总之,化学键理论是化学学科的核心,它揭示了化学键的本质及其反应机理,为探索化学反应规律和推进实用化学技术发展提供了基础。
为了更好地掌握化学反应过程,我们需要深入了解化学键理论,并将其应用于实践中。
化学键理论

化学键理论1. 引言化学键理论是化学的基础理论之一,用于解释物质中原子如何通过共用、离子、金属等键形成化合物。
本文将介绍化学键的概念、类型、强度和特点,以及相关的分子轨道理论和晶体结构中的键。
2. 化学键的概念化学键是由原子之间的相互作用力形成的,用于稳定原子之间的连接,以形成化合物。
它是化学反应和化学转化的基础。
根据原子之间电子的共享或转移方式,化学键可分为共价键、离子键和金属键三种类型。
2.1 共价键共价键是由两个原子共用一对电子而形成的。
在共价键中,原子之间的电子密度共享,以形成一个稳定的化合物。
共价键的强度取决于原子间的电子云重叠程度。
2.2 离子键离子键是由正负电荷之间的相互作用力形成的。
离子键通常存在于由金属和非金属元素组成的化合物中,其中金属原子失去电子形成阳离子,非金属原子获得电子形成阴离子。
离子键的强度取决于产生的离子之间的吸引力。
2.3 金属键金属键是金属原子之间的强电子云相互作用力形成的。
金属键的特点是原子之间的电子云重叠形成一个导电的金属电子海,这种电子海使得金属具有良好的导电性和延展性。
3. 化学键的强度和特点化学键的强度决定了化合物的稳定性和性质。
共价键通常强于离子键和金属键。
化学键的强度可以通过键能来衡量,键能是在形成化学键时放出或吸收的能量。
化学键的特点还包括键长和键角。
键长是指两个原子之间的距离,它通过实验或计算得到。
键角是指连接三个原子的两个化学键之间的夹角,它决定了分子的形状和空间结构。
4. 分子轨道理论分子轨道理论是用于描述共价键形成和分子性质的理论。
根据分子轨道理论,原子中的原子轨道会线性组合形成分子轨道。
分子轨道存在于整个分子中,描述了共价键中电子的分布情况。
常见的分子轨道包括Sigma(σ)轨道和Pi(π)轨道。
Sigma轨道是由轴向重叠形成的,是共价键中电子密度最高的轨道。
Pi轨道则是通过平面上的侧向重叠形成的,通常存在于双键和三键中。
5. 晶体结构中的键除了在分子中形成化学键外,化学键也存在于晶体结构中。
无机化学7.3化学键理论
n
价轨道数
最大成键数
2
4(2s,2px,2py,2pz)
4
3 9 (3s,3px,3py,3pz,3dz2,3dx2-y2,3dxy,3dxz,3dyz) 6
(2) 方向性
除s 轨道(角度部分为球形)外,p 、 d、f 原子轨 道在空间只有沿着一定的方向与别的原子轨道重叠, 才会产生“最大重叠”;两轨道重叠面积↑,电子在两 核间出现的几率密度↑,共价键强度↑。
键级=
成键分子轨道电子数
反键分子轨道电子数
2
20 2
1
(相当于共价单键)
2键. 级H2=+(1 氢2 0分子0.离5单子电)子键(HV2B[(无1S此)1说] 法)
3.He2 He 2 [( 1S )2 (1*S )2 ]
键级= 2 2 0 (不成键) 2
不能稳定存在
4. He2+
He2
(
1S
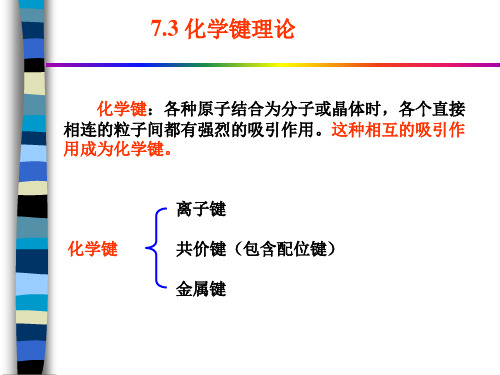
7.3 化学键理论
化学键:各种原子结合为分子或晶体时,各个直接 相连的粒子间都有强烈的吸引作用。这种相互的吸引作 用成为化学键。
化学键
离子键 共价键(包含配位键) 金属键
化学键理论
离子键理论: 共价健理论:
金属键理论:
Na+Cl-
, Ca2+O2-
H-H , H-Cl,
NN , H3C-CH3 , H2C=CH2 , HCCH Na, Mg, Al, K, Ca,
O2 , F2 , Ne2 :
E (2p) > E (2p)
第二周期元素分子轨道的形成
Li2 – N2
O2 – Ne2
第二周期同核双原子分子的分子轨道能级
Li2,Be2, B2, C2,N2 分子轨道能级顺序为:
分子结构-化学键理论概述

共价键的本质——从上面分析可知, 共价键依然是电性的,本质是轨道重 叠和电子共用,但这时是共用电子形 成的负电区域的引力,而不是库仑静 电引力。
11-2-3 现代价键理论(电子配对法) 的要点
1 共价键的形成
鲍林等人将海特勒-伦敦氢分子方法推 广到其他复杂分子系统中,认为共价键的形 成必须符合以下原理:
11-1 离子键理论
11-1-1 离子键的形成
1916年德国化学家柯塞尔(科塞尔) 根据稀有气体具有稳定结构的事实提 出了离子键理论。
离子键的形成机制
稳定倾向——活泼金属原子和活泼非金属原子接近 时,都有达到稳定稀有气体结构的倾向。
电子转移——这时活泼金属原子易失去价电子,成 为带正电荷的正离子 (阳离子);活泼非金属原子易 得到相应电子,成为带负电荷的负离子 (阴离子), 即电子转移。一旦形成正负离子,两者继续靠近时 电子结构变化不大。
d = r++r就可以得到其他离子的半径。
离子半径(pm)
离子半径变化规律
同主族上下同电荷离子半径增加; 同周期中,正离子电荷越高越小, 负离子电荷越高越大。
同元素:正离子<原子<负离子。
对性质的影响——离子半径越小,引力 越大,熔点和沸点越高(限于典型的离 子晶体)。同时影响化学性质,如I-、 Br -、Cl -、 F-的还原性依次降低。
计算——晶格能难以直接测量,可应 用玻恩-哈勃循环间接测量得到(实际
得到的是ΔH,但和ΔU相差不大,因此忽略 了差别 )。
玻恩-哈勃循环(重要) Q Na ( s ) + 1/2 F2 ( g )
S
D/2
Na ( g )
F (g)
I
E
NaF ( s ) U0
化学键理论

偶联剂分子应至少含有两种官能团,第一种官能团在理论上可于增强材料起化学反应,第二种官能团在理论上应能参与树脂的固化反应,与树脂分子链形成化学键结合,于是,偶联剂分子像“桥”一样,将增强材料与基体通过共价键牢固地连接在一起了。
1简介1949年,Bjorksten和Lyaeger共同提出化学键理论。
关于分子(或晶体)内相邻原子(或离子)间相互结合的理论。
按照这种理论,原子(或离子)是以化学键的形式结合成分子(或晶体)的。
形成化学键的物理机制是电磁相互作用。
2重要意义分子中元素原子的电子从一个原子转移到另一个原子而形成正负离子,由电荷相反的正负离子通过其过剩电荷的库伦力彼此吸引形成分子,这种静电库伦力称为离子键;原子间以共享电子对的方式形成分子,这种化学键称为共价键;在通常情况下,共价键共享的电子对分别由两个原子提供,有时共享的电子对则是由一个原子提供的,这样的共价键称为配位共价键;联结金属原子的键称为金属键,金属键的最显著特点是成键电子的流动性,它使金属表现出高度的导电性和导热性;由极性很强的化合物H-X键上的氢原子与另一个键中电负性很大的原子X上的孤立电子相互吸引而形成的分子之间的一种结合力叫氢键。
氢键不是化学键,氢键属于分子间作用力。
氢键的作用力比范德华力强而比化学键弱。
氢键在生理学和蛋白质结构化学上具有重要的意义。
3人类认识人类对物质结合方式的认识源远流长。
在古希腊,恩培多克勒用爱和恨说明物质间的结合和分离,德谟克利特则用原子的漩涡运动说明原子的聚集和分散。
中世纪的J.R.格劳伯(1604~1670)提出了物质同类相亲、异类相斥的思想。
其后还出现了关于物质结合的亲和力说,认为物质的微粒具有亲和力,由此互相吸引而结合在一起。
19世纪初,瑞典化学家J.J.贝采利乌斯(1779~1848)提出了一种建立在正负电相互吸引的观念基础上的电化二元说,从而使亲和力说更加系统化。
阐明分子中原子相互作用的经典价键理论是在原子概念基础上形成的。
化学键理论概述

波恩-哈伯循环 Na ( s ) + 1/2Cl2 ( g )
Δ f HӨm NaCl ( s )
ΔH1=ΔHvap ΔH2 =1/2 E Na ( g ) ΔH3=I1 Cl ( g ) ΔH4 =-Eea,1 ΔH5= - U
Na+( g ) + Cl-( g )
ΔfHӨm = ΔH1 + ΔH2 + ΔH3 +ΔH4 + ΔH5 = ΔHvap + 1/2E+ I1- Eea,1-U
V吸引 = -
q+ · q4 πε0 r
正、负离子之间的总势能与距离 r 关系的势能曲线。
Vp
NaCl 的势能曲线
0
Vp r0
r0
r
近距离相互排斥,远距离相互吸引, 在某一平衡距离时,吸引排斥处于动态平衡,体系势能最小,最稳定。 平衡距离 r0 —— 化学键
配位数 Na+ 6 Cl- 6 无方向性: 电荷球形对称分布 无饱和性: 空间条件允许的情况下,尽可能多的吸引相反的离子。 每个离子周围排列的异号离子的数目是一定的,实际数目与离子半 径及所带的电荷有关。
正离子和负离子之间通过静电引力结合在一起,形 成离子化合物。这种正、负离子间的静电吸引力就叫做 离子键。 当不同的原子通过离子键结合形成分子时,必然伴随 着体系能量的变化,而且新体系的能量大大低于旧体系。 根据库仑定律,两个距离为r,带有相反电荷 q+ 和 q- 的正、 负离子之间的势能 V吸引为:
缺电子体系
奇数电子体系 多电子体系
Be原子半径小,不能有大的形式电荷。
+1 -2 +1
F = Be = • F • •
•
第七章 化学键理论概述
3
BF CH BeCl 3 4 2 实例 HgCl2 BCl3 SiCl4 Be(ⅡA) B(ⅢA) C,Si 中心原子 Hg(ⅡB) (ⅣA)
PH3 N,P
(ⅤA)
NH 3
H2O H2S O,S
(ⅥA)
(5)sp3d2杂化
定义:同一原子内,由1个ns轨道与3个np轨道、 2个nd轨道间发生的杂化叫sp3d2杂化。杂化后形成 的6个新轨道叫sp3d2杂化轨道。 特点:每个sp3d2杂化轨道中含有1/6s成分和 3/6的p成分、2/6的d成分。
2.杂化轨道类型与分子的空间构型 ①sp杂化
由1个ns轨道和1个np轨道进行杂化,组成2个等同的sp杂化轨道。
每个sp杂化轨道中含1/2 s成分和1/2 p的成分。 两个sp杂化轨道之间夹角为180°,分子空间构型为直线型。
BeCl2分子形成过程
②sp2杂化
1个ns轨道和2个np轨道经杂化组成3个等同的sp2杂化轨道。
第七章
分子结构
离子键理论 化学键理论 共价键理论 金属键理论
§7 - 1 离子键理论
一、离子键的特点 1. 离子键的本质是库仑静电作用力 + f ∝ q q /r
q+、q-为离子所带电荷, R为离子间距离。
离子键强度是用晶格能来描述的。
2.离子键的特点:
既无方向性,也无饱和性。 离子化合物是由正负离子通过离子键相互交替连 结而成的晶体结构。
Na(s) + 1/2F2(g) S Na(g) I 1/2D F(g) A
ΔH
NaF(s)
U
Na+(g)
+ F-(g)
式中 S为 Na 的升华热 (108.8 kJ· mol - 1) , I 为 Na 的电离势( 495.8 kJ· mol - 1) , D 为 F 的 键 能 (141.8 kJ· mol - 1 ) , A 为 F 的 电 子 亲 合 势 (-328.0 2 kJ· mol-1),ΔH为NaF的生成焓(-573.65 kJ· mol-1),U为NaF的晶格能。
第7章 化学键理论概述
3)形成离子键, 释放能量大
在形成离子键时, 以放热的形式, 释放较 大的能量.
7-1-2
离子键的特征
1. 作用力的实质是静电引力
2. 离子键无方向性, 无饱和性 因为是静电吸引, 所以无方向性; 且 只要是正负离子之间, 则彼此吸引, 即无 饱和性.
7-1-3 离子键的强度
1. 键能和晶格能 :以 NaCl 为例: 键能:1mol 气态 NaCl 分子, 离解成气体原子时, 所吸 收的能量. 用Ei 表示: 晶格能:气态的正负离子, 结合成 1mol NaCl 晶体时, 放出的能量. 用 U 表示: 晶格能 U 越大, 则形成离子键时放出的能量越多, 离子键越强.键能和晶格能, 均能表示离子键的强度, 而且大小关系一致. 通常, 晶格能比较常用. 如何求得 晶格能?
3. 立方晶系 AB型离子晶体的空间结构 晶胞的平行六面体是正六面体时, 我们称它属于 立方晶系, 用来表示平行六面体的三度的三个轴, 称 为晶轴, 三个晶轴的长度分别用a, b, c表示, 三个晶轴 之间的夹角分别用α,β,γ表示.
我们讨论的AB型晶体指正负离子数目相同, 包括 NaCl, CsCl, ZnS.
1926年, 哥德希密特(Goldschmidt)用光学方法测定, 得到 了F-和O2-的半径, 分别为133pm 和132pm,结合X射线衍射数据, 得到一系列离子半径: 这种半径为哥德希密特半径.
1927年, Pauling 用最外层电子到核的距 离, 定义为离子半径, 并利用有效核电荷 等关系, 求出一套离子半径数据, 称为 Pauling 半径. 教材上两套数据均列出. 一般在比较 半径大小和讨论规律变化时, 多采用 Pauling 半径.
2)易形成稳定离子 Na+ 2s22p6, Cl- 3s23p6 , 达到稀有气体稳定 结构. Ag+ 4d10 轨道全充满的稳定结构. 而: C和Si 原子的电子结构为s2p2, 要失去全部 的4e, 才能形成稳定离子, 比较困难. 所以一般 不形成离子键.如 CCl4, SiF4 等, 均为共价化合 物。
《无机化学》第7章化学键理论与分子结构
《无机化学》第7章化学键理论与分子结构无机化学是研究无机物质的性质、结构和合成方法的科学。
无机化学中的化学键理论与分子结构是无机化学的重要内容之一化学键是由原子之间电子的相互作用而形成的,在无机化学中,电子主要通过离子键、共价键和金属键来相互作用。
化学键的类型取决于参与形成键的原子的电子数目和结合能力。
离子键是由阳离子和阴离子之间的静电相互作用形成的。
在化学键中,金属原子失去电子成为阳离子,非金属原子获得电子成为阴离子,从而形成的化合物具有离子晶体结构。
离子键通常具有高熔点和可溶性的特点。
共价键是由非金属原子之间的共享电子形成的。
共价键的形成过程涉及到原子间的电子云的重叠,从而共享外层电子。
共价键可以根据电子云的叠加程度分为σ键和π键。
σ键是主要的共价键,π键则是由额外的p轨道重叠形成。
在分子中,共价键的形成能够使得原子达到稳定的价电子层结构。
金属键是由金属原子之间的电子云形成的。
金属原子的价电子在整个金属晶体中自由移动,形成了金属键。
金属键的形成使得金属具有良好的导电性和热导性。
分子结构是由化学键连接在一起的原子的组合。
分子结构决定了分子的性质和反应行为。
分子结构的研究可以通过实验方法,如X射线晶体结构分析、核磁共振谱等技术,也可以通过计算化学方法进行预测和模拟。
简单分子的结构可以由初始条件和分子对称性来确定,而复杂分子的结构则需要借助实验和计算方法的综合分析。
通过对化学键理论和分子结构的研究,我们可以了解无机化合物的形成和性质,为无机化学的应用和发展提供理论基础。
此外,还可以通过对分子结构的研究来设计和合成具有特定性质和功能的无机化合物。
综上所述,化学键理论与分子结构是无机化学中的重要内容,通过研究化学键的类型和分子结构,可以揭示无机物质的性质和反应行为,并为无机化学的应用和研究提供基础。
无机化学的发展离不开对化学键理论和分子结构的深入研究。
《无机化学》第7章化学键理论与分子结构
根据共用电子对来源不同, 可分为: 一般共价键 共价键 特殊共价键(配位键)
◎配位共价键(共价键的一个特例) ◆定义:在成键的两原子中,由一方单独 提供孤对电子进入另一方的价层空轨道共 用所形成的共价键。 ◆形成条件 (1)一个原子的价电子层有孤对电子。 (2)另一个原子的价电子层有空轨道。
现代价键理论 NH3 + H+ NH4+
2 Be 的外层电子排布: 2s 4
已知实验事实: 有2个等同的Be—Cl键
键角180。
直线形分子
BeCl2分子空间构型—等性sp杂化 2p 2p
激发
2s (基态)
2s (激发态)
杂 化
2p
σ sp—p
与2个Cl的3p (化合态)轨道重叠成键
2pLeabharlann sp (杂化态)2个sp杂化轨道
BeCl2的空间构型—sp杂化
■当相互靠近的两个氢原子中的单电子自
旋方向相同时
不能成键 ψ2
排斥态 0
0
电子云稀疏区
r(pm)
排斥态:
图9-2 两个氢原子接近时的能量变化曲线(排斥态)
【例】两个H原子所处状态
n=1, l=0, m=0, ms = +1/2 可配对成键 n=1, l=0, m=0, ms = -1/2
若两个H原子的ms同为+1/2 (或-1/2), 则不能配对成键。
原子轨道杂化后,其角度分布发生 了变化。 杂化轨道的角度波函数在某个方向 的值比杂化前大得多(从角度分布图形 可看出),更有利于原子轨道间最大程 度的重叠,提高了成键能力。其大小顺 序为:
s<p<sp<sp2<sp3
杂化轨道理论
y
+ x y
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
化学键理论概述
7-1 离子键理论
1916 年德国科学家科塞尔(Kossel )提出离子键理论。
7-1-1 离子键的形成
电子转移形成离子,相应的电子构型变为稀有气体原子的电子层构型,形成稳定的离子。
正、负离子在静电引力的作用下结合在一起,形成离子化合物。
正、负离子之间的静电引力就是离子键。
r q q V 04ε -+∙-=吸引
离子间距与势能V 的变化曲线
7―1―2 离子键的性质离子键的本质是静电作用力。
离子的电荷越大,离子间的距离
越小,离子间的静电引力越强。
静电引力的实质,决定了一个离子与任何方向的电性不同的离
子相吸引而成键,所以离子键无方向性;而且只要是正负离子之间,则彼此吸引,即离子键无饱和性。
但是每个离子周围排列的相反电荷离子的数目是一定的,这个数目是与正负离子半径的大小和所带电荷多少等有关。
离子键形成的重要条件就是元素之间的电负性差值较大。
一般来说,元素的电负性差越大,形成的离子键越强。
化合物中不存在百分之百的离子键
一般用离子性百分数来表示键的离子性的相对大小。
一般认为,∆χ> 1.7,发生电子转移,主要形成离子键。
∆χ< 1.7,不发生电子转移,主要形成共价键。
7―1―3 离子键的强度键能 1 mol 气态分子,离解成气态原子时,所吸收的能量,为离子键的键能,用E i表示。
键能E i越大,表示离子键越强。
晶格能在标准状态下,将1mol 离子型晶体分解成 1 mol 气态正、负离子时需要的能量,用U表示。
晶格能U越大,表示晶体分解成离子时
吸收的能量越多,说明离子键越强。
离子键的强度通常用晶格能的大小来衡量。
所以,离子化合物中离子键力是晶体中吸引力和排斥力综合平衡的结果。
离子型化合物在通常状态下是以阴、阳离子聚集在一起形成的巨分子的形式存在。
所以离子化合物的化学结合力不是简单的两个阴、阳离子之间的结合,而是整块晶体之内的整个结合力。
因此,用晶格能描述离
子键的强度经常比离子键的键能来得更好。
玻恩 (Born )和哈伯(Haber )根据盖
斯定律建立了著名的玻恩―哈伯循环,用热
力学计算的方法解决晶格能的问题。
玻恩 (Born )和兰德(Lande )从静电引力理论出发,推导出计算晶格能的玻恩—兰德方程:
-+Z Z ,分别为正负离子的电荷数;
A 为马德隆常数,与晶格类型有关;r 为正负离子半径之和;n 为玻恩指数。
A 马德隆常数,与离子晶体结构有关:为Born 指数,它与离子的电子构型有关:
离子的电子构
型 He Ne Ar Kr Xe
n 5 7 9 10 12 卡普斯钦斯基(Kapustinskii)方程
另一种比较简单的近似式
)(10071.1130--
+-
+⋅+⨯-=mol kJ r r Z mZ U 7―1―4 离子的特征
影响晶格能的主要因素是离子的电荷、半径和电子构型。
这三个因素也是单原子离子的三个重要的特征,它们决定了离子化合物的性质。
1 离子的电荷
离子电荷高,与异号电荷间的吸引力
大,晶格能越大,离子键越强,离子化合物熔沸点高。
离子的电荷不仅影响离子化合物的物理性质,如熔点、沸点、颜色、溶解度等,而且影响离子化合物的化学性质
2 离子的电子构型
①0 电子构型如H+
② 2 电子构型(n s2)如Li+,Be2+
③8 电子构型(n s2 n p6)如Na+,Ca2+
④9~17 电子构型(916
2-
np
ns)如Fe2+,Cr3+
n
⑤18 电子构型(n s2n p6 n d10)
如Ag+,Cd2+
⑥(18 + 2)电子构型
2)1
6
2
10
n
s
n-
-
-
n
p
d
(
(
)1
(ns
)1
如Sn2+,Bi3+
离子的电子构型与离子键的强度有关,对离子化合物的性质有
响。
3 离子半径
将离子晶体中的离子看成是相切的球体,正负离子的核间距d
则是 r + 和 r - 之和
1926 年,哥德希密特(Goldschmidt )用光学方法测得 F - 和 O 2- 的半径,分别为133 pm 和 132 pm 。
结合 X 射线衍射所得的 d 值,得到一系列离子半径。
这种离子半径为哥德希密特半径。
1927 年,鲍林把最外层电子到核的距
离,定义为离子半径,并利用有效核电荷等数据,求出一套离子半径数值,被称为鲍林半径。
σ-=Z Cn
r ,式中 Z 为核电荷,σ 为屏蔽常数,C n 为由最外电子层的主量子数 n 决定的常数。
① 同一种离子,配位数大时其半径大,配位数小时其半径小。
② 同一元素的正离子半径一般小于它的原子半径,负离子半径大于它的原子半径。
正离子半径一般较小,负离子半径较大;同一元素,不同电荷数的正离子,电荷数高时,正离子半径小。
③ 同主族从上到下,电子层增加,具有相同电荷数的离子半径增大。
同周期中,正离子的电荷数越高,半径越小;负离子的电荷数越高,半径越大。
7―1―5 离子晶体
1 晶体的基本概念
⎪⎪⎪⎪⎪⎪⎩
⎪⎪⎪⎪⎪⎪⎨⎧⎪⎪⎪⎩⎪⎪⎪⎨⎧和各向异性具有固定的熔点、沸点具有规则的几何外形排列而成按一定规律周期性重复
空间由原子、分子或离子在晶体非晶体固态晶体 应用 X 射线衍射方法研究晶体结构表
明,组成晶体的结构单元在空间周期性地有
规则地排列着。
将每一个结构单元抽象成一个点,这些
点即在空间形成点阵。
用三组互不平行的直线联结空间点阵的
阵点,即形成晶格。
晶格反映晶体结构的周期性,是晶体 的数学抽象。
在晶体有规律的排列中,可以找到代 表晶体结构的最小的平行六面体单位,即
晶胞 晶胞的代表性体现在以下两个方面: 代表晶体的化学组成 代表晶体的对称性,即与晶体具有相同的对称元素
晶胞是晶体中的最小单位。
晶胞并置起来,则得到晶体。
晶胞在三维空间的无限地重复就
得到宏观的晶体。
晶胞的大小和形状由六个晶胞参
数决定。
晶胞平行六面体,始于同一顶点的三个
边,称为三个晶轴。
三个晶轴的长度分别用 a ,b ,c 表示。
三个晶轴之间的夹角分别用 α,β,γ 表示。
尽管自然界晶体有
千万种,但它们
的结构特点决定其晶胞参数只能归结为
七大类,称为七大晶系。
晶系晶轴轴间夹角
立方 a = b =cα=β =γ=90 o
四方 a = b ≠cα=β =γ=90 o
正交 a ≠b ≠cα=β =γ=90 o
三方 a = b =cα=β =γ≠90 o
六方 a = b ≠ cα=β =90 o γ=120 o
单斜 a ≠b ≠cα=β =90 o γ≠ 90 o
三斜 a ≠b ≠cα≠β≠γ≠90 o
2 离子晶体的特性
由正、负离子通过离子键结合而成的晶
体,称为离子晶体。
在离子晶体中,不存在单个分子,整个晶体可以看成是一个巨型分子,没有确定的
相对分子质量。
离子晶体中,质点之间的作用力是静电
作用力。
由于静电引力较强,晶格能较大,
所以离子晶体的熔点、沸点较高。
因离子键强度大,所以离子晶体硬度
高,但比较脆,延展性差。