化学键理论概述
化学中的化学键理论

化学中的化学键理论化学键是指原子间的吸引力力,是分子形成的基础。
化学键的形成、性质和断裂是化学反应的重要环节,也是化学研究的核心内容。
化学键理论是化学学科中的重要分支之一,它揭示了化学键的性质和本质,为化学科学的发展和应用提供了理论基础。
1. 传统化学键理论在传统的化学中,原子间的化学键是指开尔文的“亲和力”理论。
它将原子的吸引力定义为原子核和共享了某些电荷的电子间的作用力,是一个纯经验的观点。
它不是一个特别准确的预测性理论,但是仍然在一些情况下被广泛使用。
2. 共价键理论共价键理论是指两个原子通过共享电子共同发展出的化学键。
这一理论揭示了共价键的本质,即原子间电子的共享。
共价键通常用杂化轨道理论来解释。
杂化轨道理论认为,原子的价电子空壳轨道中的电子可能会混合成新的、更稳定的轨道,称为杂化轨道。
杂化轨道提供了一个更准确的方法来描述共价键——如在氨分子中,氮原子价电子空壳轨道和氢原子的原子轨道混合,产生了四个杂化轨道,分别用于和四个氢原子组成共价键。
3. 离子键理论离子键理论是指形成离子键的原理。
它是一种典型的原子或分子排斥的现象。
当两种化学物质中含有带电离子时,离子间会产生电吸引力,因此导致它们结合到一起,而这些带电离子被称为离子。
离子键通常发生在化合物中,如氯化钠(NaCl)和硫酸二钾(K2SO4)。
4. 金属键理论金属键是指金属中的化学键,通常是由金属离子通过共享电子形成金属键。
金属离子在结晶中排列成空间有序的三维结构,形成晶格。
这种排列方式为金属提供了良好的机械性能和导电性能,在大规模制造工业用金属和合金方面有着重要的应用。
总之,化学键理论是化学学科的核心,它揭示了化学键的本质及其反应机理,为探索化学反应规律和推进实用化学技术发展提供了基础。
为了更好地掌握化学反应过程,我们需要深入了解化学键理论,并将其应用于实践中。
化学键理论

化学键理论1. 引言化学键理论是化学的基础理论之一,用于解释物质中原子如何通过共用、离子、金属等键形成化合物。
本文将介绍化学键的概念、类型、强度和特点,以及相关的分子轨道理论和晶体结构中的键。
2. 化学键的概念化学键是由原子之间的相互作用力形成的,用于稳定原子之间的连接,以形成化合物。
它是化学反应和化学转化的基础。
根据原子之间电子的共享或转移方式,化学键可分为共价键、离子键和金属键三种类型。
2.1 共价键共价键是由两个原子共用一对电子而形成的。
在共价键中,原子之间的电子密度共享,以形成一个稳定的化合物。
共价键的强度取决于原子间的电子云重叠程度。
2.2 离子键离子键是由正负电荷之间的相互作用力形成的。
离子键通常存在于由金属和非金属元素组成的化合物中,其中金属原子失去电子形成阳离子,非金属原子获得电子形成阴离子。
离子键的强度取决于产生的离子之间的吸引力。
2.3 金属键金属键是金属原子之间的强电子云相互作用力形成的。
金属键的特点是原子之间的电子云重叠形成一个导电的金属电子海,这种电子海使得金属具有良好的导电性和延展性。
3. 化学键的强度和特点化学键的强度决定了化合物的稳定性和性质。
共价键通常强于离子键和金属键。
化学键的强度可以通过键能来衡量,键能是在形成化学键时放出或吸收的能量。
化学键的特点还包括键长和键角。
键长是指两个原子之间的距离,它通过实验或计算得到。
键角是指连接三个原子的两个化学键之间的夹角,它决定了分子的形状和空间结构。
4. 分子轨道理论分子轨道理论是用于描述共价键形成和分子性质的理论。
根据分子轨道理论,原子中的原子轨道会线性组合形成分子轨道。
分子轨道存在于整个分子中,描述了共价键中电子的分布情况。
常见的分子轨道包括Sigma(σ)轨道和Pi(π)轨道。
Sigma轨道是由轴向重叠形成的,是共价键中电子密度最高的轨道。
Pi轨道则是通过平面上的侧向重叠形成的,通常存在于双键和三键中。
5. 晶体结构中的键除了在分子中形成化学键外,化学键也存在于晶体结构中。
无机化学7.3化学键理论
n
价轨道数
最大成键数
2
4(2s,2px,2py,2pz)
4
3 9 (3s,3px,3py,3pz,3dz2,3dx2-y2,3dxy,3dxz,3dyz) 6
(2) 方向性
除s 轨道(角度部分为球形)外,p 、 d、f 原子轨 道在空间只有沿着一定的方向与别的原子轨道重叠, 才会产生“最大重叠”;两轨道重叠面积↑,电子在两 核间出现的几率密度↑,共价键强度↑。
键级=
成键分子轨道电子数
反键分子轨道电子数
2
20 2
1
(相当于共价单键)
2键. 级H2=+(1 氢2 0分子0.离5单子电)子键(HV2B[(无1S此)1说] 法)
3.He2 He 2 [( 1S )2 (1*S )2 ]
键级= 2 2 0 (不成键) 2
不能稳定存在
4. He2+
He2
(
1S
7.3 化学键理论
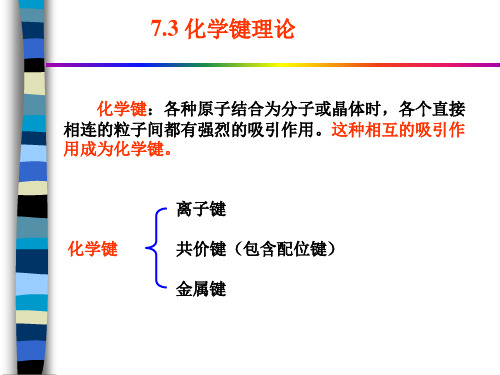
化学键:各种原子结合为分子或晶体时,各个直接 相连的粒子间都有强烈的吸引作用。这种相互的吸引作 用成为化学键。
化学键
离子键 共价键(包含配位键) 金属键
化学键理论
离子键理论: 共价健理论:
金属键理论:
Na+Cl-
, Ca2+O2-
H-H , H-Cl,
NN , H3C-CH3 , H2C=CH2 , HCCH Na, Mg, Al, K, Ca,
O2 , F2 , Ne2 :
E (2p) > E (2p)
第二周期元素分子轨道的形成
Li2 – N2
O2 – Ne2
第二周期同核双原子分子的分子轨道能级
Li2,Be2, B2, C2,N2 分子轨道能级顺序为:
化学键的价电子对排斥理论

化学键的价电子对排斥理论化学键是不同元素之间的相互作用力,让原子能够形成稳定的分子。
在化学键的形成过程中,原子的价电子对的排列有着非常重要的影响。
本文将介绍化学键的价电子对排斥理论,并探讨其在化学反应和分子结构中的应用。
一、化学键的概念与价电子对化学键是原子之间由于电子重新分配而形成的相互作用力。
原子的外层电子数目直接决定了其化学性质,而形成化学键的主要是原子的外层价电子。
原子通过与其他原子共享或转让电子来达到稳定的电子构型。
原子中的价电子对是指参与化学键形成的电子对。
对于主族元素,它们的外层电子数等于它们的主族号,即它们的电子构型为ns^2np^6。
原子需要充满外层电子轨道的电子数等于该原子主族号。
比如氧原子(O)的主族号为16,因此氧原子中的价电子对数为6。
二、价电子对排斥理论的提出价电子对排斥理论是由盖伦-赛克斯(Gillespie)和纳伊伯(Nyholm)于1957年提出的。
该理论认为,在分子中,原子上的价电子对会相互排斥,使得它们尽可能地远离彼此,以减小排斥力的作用。
这种排斥力对分子的结构和化学反应产生了重要影响。
三、价电子对排斥理论对分子几何结构的影响根据价电子对排斥理论,分子中电子对的互相排斥会导致分子的几何结构发生调整,以最大程度地降低电子对之间的排斥力。
根据电子对的排布情况,常见的几何结构可以分为线性、三角形平面、四面体、五角形平面等。
以水分子(H2O)为例,氧原子中有2对非共享的孤对电子和2对与氢原子共享的电子对。
这些电子对的排列使得水分子呈现出角度为104.5度的V型结构。
这是因为两对孤对电子通过与两个氢原子的电子云产生静电斥力,使得氢原子之间的角度变成了近似109.5度而不是预期中的120度。
四、价电子对排斥理论在分子极性和化学反应中的应用价电子对排斥理论有助于解释分子的极性和化学反应的发生。
在分子中,如果化学键中的电子对较多,则分子呈极性。
例如,二氧化碳(CO2)分子由于氧气原子周围有两对非共享电子对,因此CO2是无极性分子。
分子结构-化学键理论概述

共价键的本质——从上面分析可知, 共价键依然是电性的,本质是轨道重 叠和电子共用,但这时是共用电子形 成的负电区域的引力,而不是库仑静 电引力。
11-2-3 现代价键理论(电子配对法) 的要点
1 共价键的形成
鲍林等人将海特勒-伦敦氢分子方法推 广到其他复杂分子系统中,认为共价键的形 成必须符合以下原理:
11-1 离子键理论
11-1-1 离子键的形成
1916年德国化学家柯塞尔(科塞尔) 根据稀有气体具有稳定结构的事实提 出了离子键理论。
离子键的形成机制
稳定倾向——活泼金属原子和活泼非金属原子接近 时,都有达到稳定稀有气体结构的倾向。
电子转移——这时活泼金属原子易失去价电子,成 为带正电荷的正离子 (阳离子);活泼非金属原子易 得到相应电子,成为带负电荷的负离子 (阴离子), 即电子转移。一旦形成正负离子,两者继续靠近时 电子结构变化不大。
d = r++r就可以得到其他离子的半径。
离子半径(pm)
离子半径变化规律
同主族上下同电荷离子半径增加; 同周期中,正离子电荷越高越小, 负离子电荷越高越大。
同元素:正离子<原子<负离子。
对性质的影响——离子半径越小,引力 越大,熔点和沸点越高(限于典型的离 子晶体)。同时影响化学性质,如I-、 Br -、Cl -、 F-的还原性依次降低。
计算——晶格能难以直接测量,可应 用玻恩-哈勃循环间接测量得到(实际
得到的是ΔH,但和ΔU相差不大,因此忽略 了差别 )。
玻恩-哈勃循环(重要) Q Na ( s ) + 1/2 F2 ( g )
S
D/2
Na ( g )
F (g)
I
E
NaF ( s ) U0
化学键理论概述

波恩-哈伯循环 Na ( s ) + 1/2Cl2 ( g )
Δ f HӨm NaCl ( s )
ΔH1=ΔHvap ΔH2 =1/2 E Na ( g ) ΔH3=I1 Cl ( g ) ΔH4 =-Eea,1 ΔH5= - U
Na+( g ) + Cl-( g )
ΔfHӨm = ΔH1 + ΔH2 + ΔH3 +ΔH4 + ΔH5 = ΔHvap + 1/2E+ I1- Eea,1-U
V吸引 = -
q+ · q4 πε0 r
正、负离子之间的总势能与距离 r 关系的势能曲线。
Vp
NaCl 的势能曲线
0
Vp r0
r0
r
近距离相互排斥,远距离相互吸引, 在某一平衡距离时,吸引排斥处于动态平衡,体系势能最小,最稳定。 平衡距离 r0 —— 化学键
配位数 Na+ 6 Cl- 6 无方向性: 电荷球形对称分布 无饱和性: 空间条件允许的情况下,尽可能多的吸引相反的离子。 每个离子周围排列的异号离子的数目是一定的,实际数目与离子半 径及所带的电荷有关。
正离子和负离子之间通过静电引力结合在一起,形 成离子化合物。这种正、负离子间的静电吸引力就叫做 离子键。 当不同的原子通过离子键结合形成分子时,必然伴随 着体系能量的变化,而且新体系的能量大大低于旧体系。 根据库仑定律,两个距离为r,带有相反电荷 q+ 和 q- 的正、 负离子之间的势能 V吸引为:
缺电子体系
奇数电子体系 多电子体系
Be原子半径小,不能有大的形式电荷。
+1 -2 +1
F = Be = • F • •
•
第七章 化学键理论概述

3
BF CH BeCl 3 4 2 实例 HgCl2 BCl3 SiCl4 Be(ⅡA) B(ⅢA) C,Si 中心原子 Hg(ⅡB) (ⅣA)
PH3 N,P
(ⅤA)
NH 3
H2O H2S O,S
(ⅥA)
(5)sp3d2杂化
定义:同一原子内,由1个ns轨道与3个np轨道、 2个nd轨道间发生的杂化叫sp3d2杂化。杂化后形成 的6个新轨道叫sp3d2杂化轨道。 特点:每个sp3d2杂化轨道中含有1/6s成分和 3/6的p成分、2/6的d成分。
2.杂化轨道类型与分子的空间构型 ①sp杂化
由1个ns轨道和1个np轨道进行杂化,组成2个等同的sp杂化轨道。
每个sp杂化轨道中含1/2 s成分和1/2 p的成分。 两个sp杂化轨道之间夹角为180°,分子空间构型为直线型。
BeCl2分子形成过程
②sp2杂化
1个ns轨道和2个np轨道经杂化组成3个等同的sp2杂化轨道。
第七章
分子结构
离子键理论 化学键理论 共价键理论 金属键理论
§7 - 1 离子键理论
一、离子键的特点 1. 离子键的本质是库仑静电作用力 + f ∝ q q /r
q+、q-为离子所带电荷, R为离子间距离。
离子键强度是用晶格能来描述的。
2.离子键的特点:
既无方向性,也无饱和性。 离子化合物是由正负离子通过离子键相互交替连 结而成的晶体结构。
Na(s) + 1/2F2(g) S Na(g) I 1/2D F(g) A
ΔH
NaF(s)
U
Na+(g)
+ F-(g)
式中 S为 Na 的升华热 (108.8 kJ· mol - 1) , I 为 Na 的电离势( 495.8 kJ· mol - 1) , D 为 F 的 键 能 (141.8 kJ· mol - 1 ) , A 为 F 的 电 子 亲 合 势 (-328.0 2 kJ· mol-1),ΔH为NaF的生成焓(-573.65 kJ· mol-1),U为NaF的晶格能。
第七章化学键理论概述

化学键理论概述7-1 离子键理论1916 年德国科学家科塞尔(Kossel )提出离子键理论。
7-1-1 离子键的形成电子转移形成离子,相应的电子构型变为稀有气体原子的电子层构型,形成稳定的离子。
正、负离子在静电引力的作用下结合在一起,形成离子化合物。
正、负离子之间的静电引力就是离子键。
r q q V 04ε -+∙-=吸引离子间距与势能V 的变化曲线7―1―2 离子键的性质离子键的本质是静电作用力。
离子的电荷越大,离子间的距离越小,离子间的静电引力越强。
静电引力的实质,决定了一个离子与任何方向的电性不同的离子相吸引而成键,所以离子键无方向性;而且只要是正负离子之间,则彼此吸引,即离子键无饱和性。
但是每个离子周围排列的相反电荷离子的数目是一定的,这个数目是与正负离子半径的大小和所带电荷多少等有关。
离子键形成的重要条件就是元素之间的电负性差值较大。
一般来说,元素的电负性差越大,形成的离子键越强。
化合物中不存在百分之百的离子键一般用离子性百分数来表示键的离子性的相对大小。
一般认为,∆χ> 1.7,发生电子转移,主要形成离子键。
∆χ< 1.7,不发生电子转移,主要形成共价键。
7―1―3 离子键的强度键能 1 mol 气态分子,离解成气态原子时,所吸收的能量,为离子键的键能,用E i表示。
键能E i越大,表示离子键越强。
晶格能在标准状态下,将1mol 离子型晶体分解成 1 mol 气态正、负离子时需要的能量,用U表示。
晶格能U越大,表示晶体分解成离子时吸收的能量越多,说明离子键越强。
离子键的强度通常用晶格能的大小来衡量。
所以,离子化合物中离子键力是晶体中吸引力和排斥力综合平衡的结果。
离子型化合物在通常状态下是以阴、阳离子聚集在一起形成的巨分子的形式存在。
所以离子化合物的化学结合力不是简单的两个阴、阳离子之间的结合,而是整块晶体之内的整个结合力。
因此,用晶格能描述离子键的强度经常比离子键的键能来得更好。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第四章 化学键理论概述§4-1 价层电子对互斥理论简介(Valence Shell-Electron Pair Repulsion model, VSEPR)化学价理论的主要目标是说明分子结构,更重要的是预见分子结构。
价键理论和分子轨道理论在此方面是很有用的工具,近年来,又发展起来了一种概念上比较简单,能较好地判断和解释许多共价分子的空间构型的价层电子对互斥理论。
4-1-1 基本要点⑴共价分子中,中心原子与周围配置的原子或原子团(称之为配体)所形成的几何结构(空间构型),主要决定于中心原子价电子层中电子对的互相排斥作用,排斥力最小的结构最稳定。
例如BeF2分子中,提供的价电子为: Be2s2 F 2p x1(2p5)形成两条共价键,故中心原子价电子层中有两队电子,这两对电子只有彼此处在Be原子核相对的两侧,才能使排斥力最小。
因此BeF2 应该是直线形的 F—Be—F。
又如CH4 中 C 2s22p2 H1s1.C原子出四个电子,四个H原子各出一个电子,共8个价电子,形成四条共价键,因此,在C原子的价电子层中有四对电子,这四对电子按四面体伸向四面体的顶角排布,才能使斥力最小,因此 CH4 是四面体形的。
⑵价层中相邻电子对之间斥力大小决定于电子对之间的夹角和电子对的成键情况。
①电子对之间夹角越小斥力越大。
因此考虑排斥力时首先考虑夹角最小时电子对之间的斥力。
②考虑电子对的成键情况,有:孤电子对-孤电子对斥力>孤电子对-成键电子对>成键电子对-成键电子对。
LP-LP>LP-BP>Bp-BP.孤电子对(Lone Pair electron,LP) 成键电子对(Bond Pair electron,BP)③由于重键比单键所含电子数多,故有:叁键>双键>单键.⑶共价分子(含离子)的几何构型主要决定于中心原子的价层电子对的数目和类型(LP或BP). 中心原子价层电子对数与电子对几何结构的关系见p66表4.12。
中心原子价层电子对数与几何结构的关系⑷当分子中有双键、叁键等重键时,可将双键的两对电子和叁键的三对电子看作为一个单键的一对电子来处理。
例如CO2 分子中,碳原子和两个氧原子之间各有一个双键,将此双键作为一个单键处理,即把碳原子价层电子数看作两对,故应采取直线形结构 O= C=O。
4-1-2 判断共价分子构型的一般规则⑴确定中心原子价电子层中的电子对数①中心原子价电子层中的电子对数=(中心原子价电子数+配体提供的电子数)/2例如:BF3 B:2s22p1 F:2p x1(2p5)电子对数=(3+3×1)/2=3三对电子,电子对三顶点连线为三角形,因此,BF3 的几何构型为平面三角形。
②当被考虑对象为离子时,则有电子对数=(中心原子价电子数+配体提供的电子数-离子电荷数(带符号))/2例如:NH4+ N 2s22p3 H 1s1电子对数=(5+4×1-(+1))/2=4四对电子,顶点连线为四面体,因此,NH4+ 的几何结构为四面体。
对于AsO43-,属于无机含氧酸根,其结构较为复杂,将在下期介绍。
这里规定当氧族元素的原子作配体时,可以认为它们不提供电子。
因As:4s24p3,故电子对数=(5+4×0-(-3))/2=4即AsO43- 也呈四面体构型。
③出现奇数电子时,将此单电子也当作电子对对待如NO2,电子对数看作3,呈平面三角形分布。
电子对数=(5+2×0)/2=2.5≈3⑵根据中心原子价电子层中的电子对数,确定中心原子周围电子对分布的几何构型。
⑶绘出分子构型图 包括配体和孤电子对。
注意:若无孤电子对,电子对分布的几何构型与分子构型一致;若有孤电子对,电子对分布的几何构型则与分子构型不一致。
⑷根据成键电子对,孤电子对之间互相排斥作用的大小,确定排斥力最小的分子的稳定结构。
当有几种可能结构时,确定稳定结构的原则是电子对排斥作用最小。
确定稳定结构的步骤是:⑴绘出几种可能的构型图;⑵选出电子对之间的最小夹角,分析对比图中的各种排斥作用的种类和数目;⑶将排斥力最小的结构确定为稳定结构。
首先考虑孤电子对-孤电子对数目最少,如无法确定唯一结构,再考虑孤电子对-成键电子对数目最少,...。
例:推出ClF3 的空间构型提供的价电子数 Cl: 3s23p5 F:2p x1(2p5)①电子对数=(7+3×1)/2=5②电子空间排布构型为三角双锥③分子构型可能有三种:④90°夹角为最小夹角。
比较 a b c90°孤电子对-孤电子对 0 1 090°孤电子对-成键电子对 6 3 4√90°成键电子对-成键电子对 0 2 2由此确定(c),即T型的ClF 3 为稳定结构。
注意:由第一、二步推出的电子对数确定的只是电子对的空间排布构型,对于没有孤电子对的分子,这种构型与分子的空间构型一致;对于存在孤电子对的分子,这种构型与分子的空间构型不一致。
如NH 3,电子排布四面体,而分子呈角锥型。
电子对排布的空间构型与分子的空间构型是两个不同的概念。
4-1-3 小结⑴比较价层电子对互斥理论和杂化轨道理论,可见它们的结果很相符。
但价层电子对互斥理论只是一种定性的理论,它可以帮助我们很快确定分子的空间结构。
杂化轨道理论则还可以进一步说明化学键的形成原理,并可以计算键的相对稳定性。
⑵将中心原子价层电子对数和分子空间构型的关系总结如下:化学式 举例 分子构型 AB 3BF 3平面三角形 ①三对电子sp 2杂化AB 2SO 2角形或V形 AB 4CH 4四面体 AB 3NH 3三角锥 ②四对电子sp 3杂化 AB 2H 2O V形 AB 5PCl 5三角双锥 AB 4XeO 2F 2变形四面体,O与孤电子对斥力较小 AB 3ClF 3T形,∠FClF=87°40',孤电子对对Cl-F电子对排斥的影响. ③五对电子sp 3d杂化 AB 2XeF 2直线形,孤电子对总是处于三角形平面上. ④六对电子 AB 6SF 6八面体AB5XeOF4四方锥sp3d2杂化目前仅知此三种AB4XeF4正方形类型。
§4-2 分子间的作用力4-2-1分子的极化分子的极化是指分子在外电场的作用下,其量子轨道发生变形的过程。
例如,非极性分子在一般条件下,分子中正、负电荷的重心重合,μ=0,分子没有极性。
但是如果加上一外电场( 如放在电容器的两个平板之间),则分子受到电场影响, 核与电子云发生相对位移而使分子变形,此时两种电荷的重心分开,变成具有偶极的分子。
如左下图这就是非极性分子的极化过程,所形成的偶极叫做诱导偶极,电场愈强分子变形越厉害,诱导偶极的长度也愈长。
外电场一经除去诱导偶极也立即消失。
极性分子中已经存在着偶极,这种偶极叫做永久偶极。
但极性分子也会极化。
若将极性分子放在电场中,将有如下过程:⑴)分子将首先按电场方向排列起来,这个过程称为取向(极化);⑵另外,外电场对分子的核和电子云还会发生影响,使它们产生相对位移,使分子内正、负电荷重心更加分离(即分子变形)而产生诱导偶极,这样就增加了分子的偶极长度(如右上图)。
因此,极性分子的极化包括分子的取向与变形两步,与非极性分子的极化不同。
不仅外加电场对分子、原子、离子能产生极化作用,因极性分子和离子本身即是电场,当分子间或分子和离子间非常靠近时,也以产生极化作用。
分子极化是分子间产生作用力的重要原因。
4-2-2 分子间的作用力一般的共价化合物,除了分子内的原子间存在着强大的化学键力-共价键外,当分子和分子接近时,由于极化等原因,分子和分子之间还存在着一种较弱的作用力,称为分子间的作用力(或范德华引力)。
分子间作用力的大小,数量级约在几千焦.摩尔-1(40KJ.mol-1以下),约比化学键能小1-2个数量级,作用力的范围约3-5埃。
气体能凝聚成液体,液体结晶为固体,固体表面有吸附现象,粉末可压成片等,这些现象都证明分子间有作用力存在。
当液体汽化时需要克服液体分子间的引力,引力愈大,汽化热也愈大,液体的沸点就愈高。
例如酒精比水容易挥发,沸点较低(78℃),就是由于酒精分子间的引力小于水分子间的引力。
当固体共价化合物熔化为液体时,也需要克服分子间的引力,所以分子间作用力较大的共价化合物,将具有较高的熔点和熔化热。
因此分子间作用力是决定分子型物质的熔、沸点、汽化热、熔化热以及溶解度、表面张力、粘度等物理性质的主要因素。
分子间作用力主要包括以下几个部分:⑴ 取向力前已谈到,当极性分子互相靠近时,由于分子具有固有偶极,因此能产生取向极化。
此时,分子发生相对转动,使一个分子的正极吸引另一个偶极分子的负极,并使它们相同的极互相排斥。
这种由于永久偶极的取向而使偶极分子间互相吸引在一起的力称为取向力。
取向力其实质是微弱的静电引力的作用。
极性分子在较大距离时是杂乱无章的运动的,当十分靠近时就要发生分子的取向,使分子异极间相互吸引而成为较有规则的排列。
由此可见,由于固有偶极的存在,而有取向力的存在;固有偶极(即偶极矩μ)越大,取向力也越大。
取向力存在于极性分子之间。
⑵ 诱导力非极性分子在极性分子(或离子)的作用下可以产生诱导极化,因而产生诱导偶极。
这种诱导偶极与极性分子固有偶极之间产生的作用力称为诱导力。
例如,O2、N2等能溶于水,即包含着诱导力的作用。
当极性分子和极性分子靠近时,除了有取向力以外,分子间也可以互相极化,进一步引起彼此异极间的吸引和同极间的排斥,这样将使分子中的正电重心和负电重心再进一步远离,使固有偶极长度加大。
这种由于诱导力作用而使极性分子中偶极增加的部分也叫做诱导偶极。
因诱导作用的产生可使分子间的作用力更为加强,所以极性分子间也有诱导力存在。
诱导力的大小取决于极性分子的极性和被诱导分子的体积(即分子量)或变形性,体积越大的分子,越易发生变形。
⑶ 色散力对于非极性分子而言,由于每个分子中的核外电子和原子核都处在不停的运动之中,其核外电子和原子核常常会发生瞬间相对位移,由此在某一瞬间内可能引起正、负电荷重心的不重合,从而产生瞬时偶极。
瞬时偶极之间微弱的静电作用力叫做色散力。
由于瞬时偶极在不断地变换,因此色散力没有方向性。
注意:极性分子也存在着核外电子和原子核的瞬间相对位移,也会产生瞬时偶极。
故不仅在非极性分子之间有色散力的作用,在极性分子和非极性分子之间、在极性分子间也有色散力存在。
虽然瞬时偶极存在的时间极短,但是瞬时偶极的出现却是不断在重复,相邻的分子不断地以方向相反的瞬时偶极相互吸引,因此在它们之间始终有色散力在起作用。
(如N2、O2、H2、CO2、希有气体的液化,即是对存在色散力的证明)。
色散力是非极性分子间唯一存在的分子间作用力,但其它分子间也有这种作用力的存在。