尼莫地平的合成路线综述
尼莫地平乳注射液及制备方法(专利)

尼莫地平乳注射液及制备方法(专利)专利名称:尼莫地平乳注射液及制备方法技术领域:本发明涉及供静脉注射用的尼莫地平乳注射液及其制备方法。
背景技术:尼莫地平(Nimodipine)是第二代钙离子拮抗剂,该药优先作用于小动脉,选择性作用于脑血管,可明显减轻脑出血损伤并促进神经功能的恢复,临床上用于缺血性脑血管疾病、偏头痛、突发性耳聋、蛛网膜下脑出血和高血压等疾病的治疗,取得了良好的治疗效果。
尼莫地平是一种溶于乙醇、乙醚等有机溶媒而不溶于水的难溶性药物。
目前市面上销售的尼莫地平注射液均制成非水溶媒注射液,内含大量的乙醇(20~25%)和聚乙二醇(17~20%)作溶媒。
该制剂在临床应用时,主要存在浓度低(0.2mg/ml),稀释时易析出结晶以及非水溶媒对血管刺激而产生静脉炎的问题,另在临床使用时,当用5%葡萄糖注射液稀释后2~3小时,即出现针状或柱状尼莫地平结晶,含量急速下降直接影响用药安全和有效性。
为了防止稀释后的结晶析出,国外产品采用输液三通阀在线混合后立即输注,其滴注速度和剂量不易控制。
国内有些产品采用添加聚乙烯吡咯烷酮(PVP)延长结晶析出时间,其效果一般,而且由于PVP进入体内会阻碍凝血,已被国家食品和药品监督管理局(SFDA)限止使用。
本发明人发明了一种新的尼莫地平乳注射液。
发明内容本发明的目的在于提供一种尼莫地平乳注射液。
将活性成分尼莫地平加辅料注射用油、乳化剂、助溶剂、等渗调节剂及注射用水制成尼莫地平乳注射液。
由于我们在制剂中加入了适宜的助溶剂从而提高了尼莫地平在乳剂中的溶解度及制剂稳定性,使我们的尼莫地平乳剂的浓度相对较高,例如在高浓度如每ml乳注射液含1mg尼莫地平的情况下仍保持稳定。
该乳剂可直接用葡萄糖注射液稀释后使用,使用过程中未见含量下降及析出结晶。
并且每ml注射液含1mg尼莫地平的乳注射液,相对于现有技术当使用一定量的活性物质时,注射的液体大大减少,减少了病人的代谢负担。
本发明的尼莫地平乳注射液不使用乙醇、聚乙二醇等作为溶媒,减轻了对血管的刺激,保证了临床用药安全,并提高了病人用药的顺应性。
尼莫地平的合成
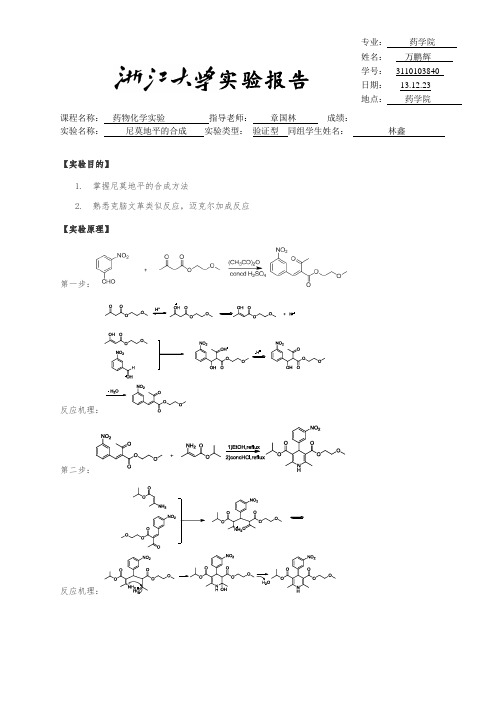
课程名称:药物化学实验指导老师:章国林成绩:__________________ 实验名称:尼莫地平的合成实验类型:_验证型同组学生姓名:林鑫【实验目的】1.掌握尼莫地平的合成方法2.熟悉克脑文革类似反应,迈克尔加成反应【实验原理】第一步:反应机理:第二步:反应机理:第一步反应:【实验材料】实验试剂:1、乙酰乙酸甲氧基乙酯9.6g (0.06mol)2、醋酸酐8.1g (密度1.08) 8ml3、浓硫酸3.2g(密度1.84)1.8ml4、间硝基苯甲醛 6.04g (0.04 mol)5、95%乙醇8mL实验仪器:1、100ml 三颈烧瓶2、10ml量筒3、布氏漏斗4、抽滤瓶5、搅拌子6、温度计7、玻璃塞8、滴液漏斗9、干燥器10、50ml烧杯11、油浴锅【实验步骤】1、在三颈烧瓶里依次加入9.6g 乙酰乙酸甲氧乙酯,8ml 醋酸酐;2、三颈烧瓶上装备1个搅拌子、1个温度计、1个塞着棉花和无水氯化钙的干燥器、1个滴液漏斗;3、置于冰水浴中冷却,搅拌下缓慢滴加浓硫酸(3.2g),(实际操作过程中,旋塞旋转过快,10s内浓硫酸已滴加完毕,升温过快,导致的影响于实验讨论中分析);4、浓硫酸滴加完毕后,加入间硝基苯甲醛(6.04g)后升至室温;5、固体溶解后搅拌反应2h,反应过程中溶液颜色逐渐变深,由最初的淡黄色变为最终的橙黄色;6、反应接近完全时,于硅胶板上点上:原料间硝基苯甲醛斑点、反应溶液斑点及两者混合斑点,确认已有产物生成;7、加入95 %乙醇( 8mL ) 搅匀,冰浴冷却,减压抽滤,得黄色滤饼;8、滤饼先用一定量95%冷乙醇洗涤,黄色褪去;9、再用一定量纯水洗涤,洗出白色絮状沉淀;10、称量滤饼湿重,放置一星期后称量干重,并计算产率。
【实验结果】1、第一步得到产物湿重 5.72g,干重为 3.10 g,产率3.10*100% 0.04*293.2726.4 %;2、从左到右依次是原料点、反应液点、混合点,明显看出有产物生成,且含量相对较多,以致拖尾;Figure 1 点板结果第二步反应【实验材料】实验试剂:1、2- ( 3- 硝基亚苄基)乙酰乙酸2- 甲氧基乙酯(第一步反应产物) 3.10g 0.0106mol2、3-氨基丁烯酸异丙酯 2.25g 0.016mol3、无水乙醇12mL4、浓盐酸 1.06mL 实验仪器:1、100ml 圆底烧瓶2、20ml量筒3、布氏漏斗4、抽滤瓶5、搅拌子6、温度计7、玻璃塞8、滴液漏斗9、干燥器10、冷凝管11、橡皮管12、充满氮气的气球13、三通活塞等【实验步骤】1、在圆底烧瓶里依次加入3.10g第一步反应产物,3-氨基巴豆酸异丙酯2.25g,无水乙醇12mL;2、三颈烧瓶上装备1个搅拌子、1根一端装有三通活塞和气球的冷凝管;3、置于油浴锅中,加热回流1小时(回流温度保持在80-90℃),反应完全后加入1.06ml浓盐酸;4、浓盐酸滴加完毕后,继续冷凝回流0.5h;5、反应停止,加入6mL水,冰浴冷却,黄色固体析出,待产物析出完全,减压抽滤;6、滤渣用50%冰乙醇洗涤,得到黄色晶体,干燥产物,称量并计算产率。
尼莫地平PLGA微球的制备方法改良及其特性研究

摘要尼莫地平(nimodipine,NMP)在临床上常被用为选择性作用于脑血管平滑肌的钙拮抗剂,己成为治疗脑血管疾病的首选药物。
针对其体内半衰期短,口服生物利用度低,须频繁给药,且副作用较严重等问题,本文将其微囊化以提高NMP的临床应用价值。
研究利用乳化-溶剂挥发法将NMP进行微囊化,载体材料采用生物可降解材料聚乳酸-羟基乙酸共聚物(PLGA)。
为了通过易挥发潜溶剂的使用来调节微球的固化速率,在油相溶剂中引入高挥发度的石油醚与二氯甲烷(DCM)作为混合溶剂,考察了石油醚与DCM的混合比例对载药微球的影响;另外考察了不同乳化剂、聚合物分子量、聚合物浓度、投药比等因素,并进行了正交优化设计。
研究还考察了减压条件下微球的制备,并与常压下优化所得的微球进行了比较。
另外,对载药微球进行了初步稳定性考察。
结果表明,聚合物分子量增大及浓度的提高均会提高油相的粘度从而增大微球的平均粒径,载药量,并降低突释率;投药比超过1:15时会在微球表面产生:V DCM = 0、1:10、药物结晶且突释率增大;随着油相中石油醚比例的增大(V石油醚1:8、1:4、1:2),微球的固化速率有不同程度的增大,从而影响微球的载药能力与体外释放等特性。
正交优化后,微球包封率(EE)相对于优化前提高52.2%,突释率相对降低58.8%,所得微球粒度分布均匀,外表光滑致密,缓释效果增强(25 d后累积释药率约80%),体外释药动力学研究结果表明药物释放遵循扩散和骨架溶蚀共同控制的机制,且工艺重现性良好;优化组载药微球与以DCM为单一溶剂制得的微球相比,包封率有小幅提高,形态改善显著;另外,石油醚的使用未改变药物与聚合物的物理存在状态,而在较小程度上降低了聚合物的玻璃化转变温度(T g)。
利用减压去除溶剂法,所得微球包封率较常压下高(EE>50%),但因其表面的少量结晶,体外释放速率较快(18 d时累积释药率即可达到80%左右)。
NMP微球初步稳定性试验结果表明,微球对湿、热及强光不稳定,宜在低温,低湿及避光条件下保存。
尼莫地平乳注射液及制备方法(专利)

尼莫地平乳注射液及制备方法(专利)尼莫地平乳注射液是一种用于治疗高血压或冠心病等心血管疾病的药物。
它主要成分是尼莫地平,属于钙通道阻滞剂。
尼莫地平能够通过阻断心血管平滑肌细胞上的L型钙通道,减少钙离子内流,从而降低心血管张力,扩张血管,降低血压,增加心肌供血,改善心血管疾病症状。
制备尼莫地平乳注射液的方法可以通过以下步骤来进行:
1.准备所需原料和药品:尼莫地平草酸盐、辅料等。
2.称取适量的尼莫地平草酸盐和辅料,按照一定的比例混合在一起。
3.在适当的容器中,加入一定量的溶剂(如无水乙醇)。
4.将混合好的尼莫地平草酸盐和辅料加入溶剂中,充分搅拌直至完全溶解。
5.对混合后的溶液进行过滤,以去除其中的不溶性杂质。
6.将过滤后的溶液转移至符合卫生标准的容器中。
7.将容器密封,并进行灭菌处理,以保证制剂的无菌性。
总结而言,尼莫地平乳注射液是一种用于治疗高血压或冠心病等心血管疾病的药物。
其制备方法包括称取适量的尼莫地平草酸盐和辅料,将其加入溶剂中并充分溶解,过滤并灭菌处理后即可得到乳注射液制剂。
这种制剂能够快速进入血液循环,发挥治疗作用。
钙拮抗剂尼群地平的合成新工艺

钙拮抗剂尼群地平的合成新工艺张邦乐何炜李晓晔朱玉屏【关键词】钙拮抗剂关键词:钙拮抗剂;1,4-二氢吡啶类化合物;尼群地平;合成0引言二氢吡啶类钙拮抗剂是钙拮抗剂中研究最活跃,近几年来新品种上市最多的一类[1].尼群地平(Nitrendipine,1)属第二代二氢吡啶类钙拮抗剂,要紧用于高血压和冠心病的医治,我国已将其审批列入国家药典并投放市场.咱们以双乙烯酮为原料,采纳两个“一锅煮”,经文献报导的改良Hantzsch吡啶合成法[2-4]进行了尼群地平的合成研究.1实验资料实验所用试剂除二氧六环(CP级)、三乙胺(CP级)、浓硫酸(AR级)外,均为工业品.TJ-27030红外光谱仪;XRC-显微熔点测定仪;UV-330型紫外可见光光度计.1.1合成线路1.2制备进程1.2.13-氨基-2-丁烯酸甲酯(3)250mL三颈瓶中加入甲醇16g,三乙胺0.1mL,搅拌混匀,水浴加热到75℃,然后慢慢滴加双乙烯酮42g.约需1.5h,反映温度操纵在80℃℃左右搅拌3~4h(薄层层析检测终点).然后将上述反映混合液冰浴冷却至0~3℃,搅拌下通入氨气反映并维持内温10℃左右.反映7h后冷却静置.待大量结晶析出后快速抽滤,洗涤得白色固体(3)46.7g.mp(熔点)83~84℃(文献82~83℃),总收率81%.1.2.23-硝基亚苄基乙酰乙酸乙酯(5)250mL三颈瓶中加入乙醇23g,三乙胺0.1mL,混匀,搅拌下水浴加热到75℃,慢慢滴加双乙烯酮42g.约需1~1.5h,反映温度始终维持80℃℃℃,搅拌下缓慢滴加浓硫酸10mL,温度操纵在3℃以下.滴加完后加入干燥磨细的间硝基苯甲醛76g,搅拌反映8h(反映温度操纵于10±2℃).静置,抽滤,水和冰乙醇洗涤,得白色晶体93g.mp(熔点)107~109℃(文献[3]mp108~109℃),两步总收率70.7%. 1.2.3(±)2,6-二甲基-4-(3-硝基苯基)-1,4-二氢-3,5-吡啶二甲酸甲乙酯(尼群地平,1)100mL圆底烧瓶中加入7.8g化合物5,3.6g化合物3和30mL二氧六环,搅拌下升℃(文献[3]158~159℃,产率:81%;药典标准mp156~159℃).其辨别、薄层层析、紫外吸收、含量测定等项目均达国家药典标准.2讨论该工艺线路操作简便,合成中间体酯2,4不经减压蒸馏纯化直接用于下步反映,在含酯2的母液中通氨后静置使3以结晶析出,幸免了文献减压蒸馏、萃取等繁琐苛刻的后处置条件;合成5时将文献报导的硫酸催化代替氯化氢引入生产环节,从而使整个进程条件温和,工序缩短,降低了本钱并减少了环境污染;环化反映中以二氧六环代替乙醇做溶剂,提高了产率且缩短了反映时刻.该线路所得产品维持三步法优势,一样易于精制,放大实验后收率稳固,质量达国家药典(一九九五年版)标准,是一种适用可行的合成方式.参考文献:[1]翁玲玲,郭丽.国外正在研制的钙拮抗剂[J].中国药学杂志,1995;30(5):303-305.[2]MeyerVH.Nimodipine:synthesisandmetabolicpathway [J].ArzneimForsch/DrugRes,1983;33(1):106-108.[3]ZhangQN,JiaXM,ZhangEQ.ImprovedsynthesisofNi-trendipine[J].ChinJPharm,1992;23(4):154-155.[4]陈新.二氢吡啶类钙拮抗剂的合成工艺研究[J].药学进展,1990;14(3):145-149.。
钙通道拮抗剂尼莫地平的合成

钙通道拮抗剂尼莫地平的合成
程霄云;高孟林
【期刊名称】《山西化工》
【年(卷),期】1995(000)004
【摘要】尼莫地平的合成以双乙烯酮(DK)、乙二醇独甲醇,异丙醇,间硝基
苯甲醛为基本原料,经缩合,氨化,Michael加成反应,环合等反应制得。
反应条件温和,副反应少,通过对少Michael加成和环合等反应进行工艺改进,使环合一步的收主经为83.72%,较文献收率高2.72%。
【总页数】2页(P28-29)
【作者】程霄云;高孟林
【作者单位】不详;不详
【正文语种】中文
【中图分类】R972
【相关文献】
1.1,4-二氢吡啶类钙拮抗剂的合成法研究(Ⅱ)尼莫地平的合成新方法 [J], 陈芬儿;
李艳;沈怡
2.二氢吡啶类钙通道拮抗剂抗心肌肥厚作用依赖于其对N-型钙通道的阻断能力 [J], 罗琼;禢婉玲;席芳;廖禹林;北风政史
3.血小板GPⅡb/Ⅲa拮抗剂RGDS和钙通道拮抗剂Nifedipine对GPIIb/IIIa活化的影响 [J], 杨国雷;尹松梅;张复华;牛国敏;凌奕文
4.钙拮抗剂尼莫地平的合成 [J], 陈新;高亚平;黄文龙;郭晓东
5.钙通道拮抗剂和钙调素拮抗剂对白血病耐药细胞的耐药基因和细胞内Ca^(2+)浓度的影响 [J], 陈同辛;沈蕾;林梓;王耀平;应大明
因版权原因,仅展示原文概要,查看原文内容请购买。
钙拮抗剂尼莫地平的合成
钙拮抗剂尼莫地平的合成
陈新;高亚平;黄文龙;郭晓东
【期刊名称】《中国医药工业杂志》
【年(卷),期】1989(20)2
【摘要】尼莫地平(1),化学名为(±)-1,4-二氢-2,6-二甲基-4-(3-硝基苯基)吡啶-3,5-二羧酸异丙酯2-甲氧基乙酯,属第二代二氢吡啶类钙拮抗剂。
它选择性地作用于脑血管,可用于预防和治疗蛛网膜下出血后脑血管痉挛所致的脑缺血、神经功能不足、痴呆、癫痫、突发性耳聋,以及脑血管机能不全等症,对偏头痛也有较好的疗效。
联邦德国于1985年5月上市,国内尚未见试制报道。
【总页数】2页(P52-53)
【关键词】尼莫地平;钙拮抗剂;合成;脑血管
【作者】陈新;高亚平;黄文龙;郭晓东
【作者单位】中国药科大学;江苏省盱眙制药厂
【正文语种】中文
【中图分类】TQ463.54
【相关文献】
1.1,4-二氢吡啶类钙拮抗剂的合成法研究(Ⅱ)尼莫地平的合成新方法 [J], 陈芬儿;李艳;沈怡
2.钙拮抗剂尼莫地平救治重型颅脑损伤的临床研究 [J], 徐如祥
3.钙拮抗剂:尼莫地平改善内皮功能 [J],
4.钙拮抗剂尼莫地平救治重型颅脑损伤的临床研究 [J], 徐如祥;陈长才;杨俊;李铁林;邓国鼎;张是宏;李新运;柯以铨;张世忠
5.钙拮抗剂尼莫地平对小鼠开场行为和学习记忆的影响 [J], 郭丹;杜红燕;薛龙增因版权原因,仅展示原文概要,查看原文内容请购买。
实验十三尼莫地平片剂的制备及检测
尼莫地平片剂的制备及检 测
一、实验目的
• 通过设计尼莫地平片剂处方和制备工艺, 并进行其片剂制备和质量检查,掌握片 剂处方及工艺设计的基本思路,探讨片 剂成型的机理及影响压片因素,熟悉片 剂的质量检查,培养学生综合设计能力 和将理论知识运用于科研、生产实பைடு நூலகம்的 能力,为研制开发药物新制剂和新剂型 奠定基础。
五、思考题
• 1. 片剂处方和工艺设计的基本思路。 • 2.怎样提高难溶性药物的溶出度?
二、实验原理
• 尼莫地平淡黄色结晶性粉末,遇光不稳 定;无臭,无味。在丙酮、氯仿中易溶, 在乙醇溶解,在乙醚中微溶,在水中不 溶。熔点为124~128℃。根据尼莫地平 的物理和化学性质制备片剂,并检测相 关指标。
三、 实验仪器与材料
• 材料:尼莫地平、辅料(自选)。
• 仪器:从单冲压片机,片剂四用仪,分 析天平,普通天平,烘箱,电炉,药筛 (80目、120目),尼龙筛(14目、16 目),搪瓷盘,乳钵自选。
四、实验内容
• 1、查阅有关尼莫地平的理化性质等文献 资料。
• 2、设计规格为20mg/片尼莫地平片剂 的处方和制备工艺、质量控制方法、稳 定性研究实验方案。
• 3、交流设计思路,在实验指导教师指导 和共同商讨下完善实验设计,经老师批 准认同后正式实施。
• 4. 完成实验并写出实验报告后,进行实 验总结和讨论。
28尼莫地平原研处方工艺分析
1.概述尼莫地平最早由德国拜耳公司以尼莫地平片(Nimotop)上市,为原研制剂。
早期用于治疗高血压治疗,现在研究指出用于预防蛛网膜下腔出血有良好效果。
本品为1,4-二氢吡啶类钙离子拮抗剂,对脑组织受体有高度选择容易透过血脑屏障。
通过有效地阻止钙离子进入细胞内、抑制平滑肌收缩,达到解除血管痉挛之目的,从而保护了脑神经元,稳定其功能及增进脑血灌流,改善脑供血,提高对缺氧的耐受力。
本品能有效地预防和治疗因蛛网膜下腔出血引起的脑血管痉挛所造成的脑组织缺血性损伤。
能降低红细胞脆性及血液黏稠度,抑制血小板聚集,抗血栓形成。
在适宜剂量下选择性扩张脑血管,几乎不影响外周血管。
本品还可以改善老年性脑损伤患者的记忆障碍。
最新循证医学结果证明本品能有效改善卒中后认知功能。
本品口服吸收快,生物利用度仅为13%,约于1小时内达到峰值浓度,半衰期约为1?2小时,彻底消除时间约为8?9小时,95%以上的药物与血浆蛋白结合。
大部分以代谢产物的形式排出体外。
本品可透过胎盘屏障,并可分泌入乳汁。
慢性肝功能损害患者中本品的生物活性增加,其血药浓度峰值可达正常人的2倍。
①用于急性脑血管病恢复期的血液循环改善。
各种原因的蛛网膜下腔出血后的脑血管痉挛,及其所致的缺血性神经障碍高血压、偏头痛等。
②也被用作缺血性神经元保护和血管性痴呆的治疗。
③对突发性耳聋也有一定疗效。
本品为浅黄色粉末,不溶于水,溶于乙醇和氯仿。
对光敏感,对热、潮湿、氧和水都较敏感。
配成的溶液暴露在光下时,很快进行光氧化而生成无活性的吡啶类化合物。
本品为光学异构体的混合物,右旋尼莫地平的作用比左旋光学异构体明显降低。
原料药性质解离常数:pKa= 5.41(强碱)在各pH值溶出介质中的溶解度(37℃):pH1.2:未测定pH4.0:未测定pH6.8:未测定水:0.012mg/ml在各溶出介质中的稳定性:水:未测定。
在各pH值溶出介质中:未测定。
光:未测定。
BCS分类:Ⅱ类2.上市情况(1)尼莫地平片:国内尼莫地平片共有67个批准文号,有三种规格:20mg、30mg和50mg。
尼莫地平的合成路线
尼莫地平(Nimodipine)的合成尼莫地平是一种Ca2+通道阻滞剂,属双氢吡啶类钙拮抗剂,容易通过血脑屏障而作用于脑血管及神经细胞。
药理特性是选择性扩张脑血管,而无盗血现象,在增加脑血流量的同时而不影响脑代谢。
可拮抗K+、5-HT、花生四烯酸,过氧化氢、TXA2、DGF2a和蛛网膜下腔出血所致脑血管痉挛。
有向精神性作用即抗抑郁和改善学习、记忆功能。
另有降低红血球脆性,血浆粘稠性和抑制血小板聚集作用。
尼莫地平片临床用于预防和治疗由于蛛网膜下腔出血后脑血管痉挛引起的缺血性神经损伤以及老年性脑功能损伤、偏头痛、突发性耳聋等。
结构式如下:合成法:第一步:缩合反应将间硝基苯甲醛( 100 g ,0 . 66 mol) ,乙酰乙酸甲氧基乙酯( 112 g ,0 . 70 mol) ,甲苯( 200 ml ) 置于1000 ml 三颈瓶中,控制温度5060 ℃,搅拌下滴入POCl 3( 25 ml ,0 . 37 mol ) ,回流4 h ,回收甲苯,残留物用95 %乙醇重结晶,得白色结晶( 17616g ,91 %) mp 6668 ℃。
经典合成法中,缩合催化剂有氯化氢气体和浓硫酸,在生产上使用氯化氢气体有三个缺点:设备腐蚀严重,氯化氢量不易控制,环境污染严重等。
采用浓硫酸做催化剂虽然没有上述缺点,但由于生产过程中采用了较大量的浓硫酸(又做溶剂) ,造成后处理困难。
鉴于此,我们选用了POCl3 做催化剂,克服了氯化氢和浓硫酸做催化剂的缺点,反应易控制,副产物少,后处理简单且产品质量好。
第二步:环合反应将 2 ( 100 g ,0 . 34 mol) ,β-氨基巴豆酸异丙酯( 50 g ,0 . 35 mol) ,二氧六环( 200 ml ) 置于500 ml三颈瓶中,加热回流1 h 后,回收二氧六环,残留物用无水乙醇重结晶,得淡黄色固体。
另外,查阅文献,得另外合成法,如来自Pharmaceutical Chemistry Journal,46(5), 285-287; 2012、Faming Zhuanli Shenqing, 102174012,07 Sep 2011等的尼莫地平合成法,都因反应时间过长、步骤过于繁琐而没有被本组采纳。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
尼莫地平的合成
尼莫地平是德国拜耳公司开发的二氢吡啶类钙拮抗剂,该药物对于治疗各种原因引起的蛛网膜下隙出血后的脑血管痉挛和改善脑血管恢复期的血液循环有着很好效果。
因此,在过去的近三十里,关于该药物合成路线的优化提高一直没有停止过。
到目前为止,尼莫地平的合成路线主要有以下几种:
1.拜耳公司的原始合成路线[1]:
1985年,拜耳采用了如下的路线合成了尼莫地平:A. 首先,以异丙醇做溶剂,乙酰乙酸2-甲氧基乙基酯(1)与间硝基苯甲醛(2)在冰醋酸/哌啶的催化下缩合生成2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)。
B. 然后2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)与3-氨基-2-丁烯酸异丙酯(4)在异丙醇中进行环化反应得到尼莫地平化合物。
2.山东新华制药厂的工艺优化[2]:
在上世纪八十年开始,山东新华制药厂对尼莫地平的合成路线进行了不断的优化。
1988年,他们在化合物A 和化合物B的环化反应中,不使用溶剂(两种化合物直接在熔融状态下)或使用环己烷做溶剂,是反应从体系中迅速
分离,反应时间短,尼莫地平的收率最高达88.56%;缺点是反应温度高,而环己烷对于两种化合物的溶解性很差,几乎也是在熔融状态下反应。
1992年,他们在该步反应中采用乙醇/环己烷为混合溶剂(V乙醇/V环己烷= 1/4),产率达到89.21%,而杂质含量小于0.2%。
2011年他们对化合物B的合成工艺也进行了优化,反应后先用CaCl2干燥除水,然后蒸馏,其收率达到81.5%,纯度达98.8%。
环化反应中采用异丙醇/环己烷作为反应溶剂,尼莫地平收率为78.8%,杂质含量0.74%。
3.
1994年,Burgurs保护了一条新的合成路线[3]:第一步(a), 乙酰乙酸异丙酯(6)与3-硝基苯甲醛(2)在异丙醇中,以冰醋酸/哌啶为催化剂进行缩合反应,生成2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(7); 第二步(b), 乙酰乙酸2-甲氧基乙基酯(1)与醋酸铵在绝对乙醇中回流生成3-氨基-2-丁烯酸2-甲氧基乙基酯(8); 第三步(c), 2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(7)和3-氨基-2-丁烯酸2-甲氧基乙基酯(8)与铝粉先微波下活化15分钟,或100摄氏度下活化2小时,然后将化合物悬浮与了二氯甲烷中,后将二氯甲烷旋干,100摄氏度下再反应30分钟。
经后处理结晶可得纯尼莫地平,收率71%。
该方法提出用醋酸铵代替氨气方法合成3-氨基-2-丁烯酸2-甲氧基乙基酯(8),操作相对简单,但第三部环化反应操作复杂,收率较低。
4.固相合成法
1996年,Gordeev等报道了一种固相合成尼莫地平的方法[4]:A.首先Fmoc保护的Rink树脂用10%哌啶的DMF 溶液脱保护,抽滤,洗涤,真空干燥之后,紧接着加入二氯甲烷,乙酰乙酸异丙酯(6)和4A分子筛,室温下搅拌三天,得到化合物10。
B.然后化合物10,乙酰乙酸2-甲氧基乙基酯(1),3-硝基苯甲醛(2)和4A分子筛在干燥的吡啶中,
45摄氏度搅拌2小时,得到化合物11。
C.化合物11用95%三氟乙酸的THF溶液或3%三氟乙酸的二氯甲烷溶液处理,脱去树脂,便得到尼莫地平,收率为78%。
得到的尼莫地平化合物仍需柱分离提纯。
该方法采用固相合成,中间产物分离简单,大大缩短提纯步骤;但固相反应载体树脂价格昂贵,不适合大批量制备。
5.
同样在1996年,Perego等人采用了对甲氧基苯甲酸/二甲胺为催化剂(0.01-0.06当量)[5],来替代拜耳公司采用的冰醋酸/哌啶催化剂,在第一步缩合反应中,2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)的收率有了一定的提高(95%收率)。
值得一提的是,当在第二部环化反应中也采用该催化剂(0.005-0.015当量),反应只需回流12小时,尼莫地平的收率便可达96%。
该方法采用对甲氧基苯甲酸/二甲胺为催化剂,降低反应温度,从而降低副产物的生成,收率有了明显的提高;但该催化体系不如冰醋酸/哌啶便宜易得。
6.
2005年,中国药科大学徐云根等人报道了一种新的催化体系[6]。
A.在乙酰乙酸2-甲氧基乙基酯(1) (过量)与间硝基苯甲醛(2)的缩合反应中,采用乙酸酐/浓硫酸的催化体系,2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)的收率为88.3%;B.在2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)与3-氨基-2-丁烯酸异丙酯(4)的缩合反应中先在乙醇中回流一小时,然后加入浓盐酸作为催化剂,回流0.5小时,尼莫地平的收率为90.8%。
该方法第一步采用乙酸酐/浓硫酸的催化体系以及第二步的浓盐酸催化体系,大大缩短了反应时间;但采用强酸对设备腐蚀大。
7.
2009年,武汉中孚生物科技有限公司以化合物12为起始原料,在阳离子交换树脂的催化下,实现了与乙二醇单甲醚(13)的酸酯缩合反应,得到尼莫地平,收率为87%,纯度达99.0%,杂质含量0.6%。
但该专利采用阳离子交换树脂做催化剂,操作简单,催化剂可再生;但起未提及化合物12的合成方法[7]。
8.
2010年天津市中央药业有限公司对尼莫地平合成路线中的第二步环化反应进行了研究,发现才用环状溶剂作为环化缩合实际,可以有效地控制终产品尼莫地平的杂质含量[8]。
采用该方法后,终产品的杂志含量可以控制在较低水平,但收率也有所降低。
9.
2012年,balaev等报道了其尼莫地平的工艺改进路线[9]:a) 首先用乙酰乙酸异丙酯(6)与氨气在异丙醇中,2-5摄氏度反应8小时,简单的蒸馏后,残留物(4)直接用于下一步反应; b) 等当量的乙酰乙酸2-甲氧基乙基酯(1)与3-硝基苯甲醛溶于甲苯中,通氯化氢气体使体系饱和,该体系在0-4摄氏度下反应24小时生成2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(3)。
在简单的洗涤,蒸馏,甲苯洗涤,干燥之后直接用于下一步反应;c) 化合物(3) (1.2当量)与3-硝基苯甲醛(1当量)在异丙醇中回流2小时,然后加入浓盐酸继续回流30分钟。
简单的结晶洗涤之后尼莫地平的收率达到91%,纯度达99.18%。
该方法中间体纯化简单,其产品收率高,纯度好,但反应过程对温度控制要求严格。
10.
2013年,MacDonald等提出了另外的合成工艺[10]:首先,将乙酰乙酸甲氧基乙基酯(1)和羟胺溶液混合得到3-氨基正丁酸甲氧基以及酯(14);然后在正丁酸/哌啶的催化下,乙酰乙酸异丙酯和3-硝基苯甲醛反应缩合生成2-(3-硝基亚苄基)乙酰乙酸2-甲氧基乙基酯(7);最后,化合物(7)和化合物(14)在正丁酸/哌啶的催化下环化生成终产物尼莫地平。
但是专利中没有给出具体的反应实例,也没有化合物收率和纯度的数据。
Reference
1.Bayer. BE 900511 A1 19850306.
2.a) Xiao, Wen-Kai. CN 1037146 A 19891115. b) Le, Wei-Pei; Gao, Guo-Feng. CN 1063684 A 19920819. c) Le, Wei-Pei;
Gao, Guo-Feng. CN 1037178 C 19980128. d) Wang, Bing; Zhang, Bo; Shen, Ping; Wang, Long-Chuang. CN 102174012 A 10110907.
3.Burgos Garcia, Carolina; Izquierdo Ceinos, Maria Luisa; Garcia Navio, Jose Luis; Vaquero Lopez, Juan Jose; Alvarez-Builla
Gomez, Julio; Novella Robisco, Jose Luis; Calatayud Arinero, Jose; Montoro Jimenez, Antonio F. ES 2055653 A1 19940816.
4.a) Gordeev, Mikhail F.; Patel, Dinesh V.; Gordon, Eric M. Journal of Organic Chemistry (1996), 61(3), 924-8. b) Gordeev,
Mikhail F.; Patel, Dinesh V. WO 9633972 A1 19961031.
5.Perego, Bruno; Manghisi, Elso. WO 9629310 A1 1996092
6.
6.Xu, Yungen; Hua, Weiyi. Zhongguo Yiyao Gongye Zazhi (2005), 36(1), 8-9.
7.Lin, Xuxing; Wu, Yunfei; Yang, Zhendong. CN 101508672 A 20090819.
8.Liu, Hui; Xu, Zhenyan; Zhang, Sha. CN 101851192 A 20101006.
9.Balaev, A. N.; Osipov, V. N.; Fedorov, V. E. Pharmaceutical Chemistry Journal (2012), 46(5), 285-287.
10.a) MacDonald, R. Loch; Davis, Cara R.; Burton, Kevin; Winchester, Gary; Stella, Angela R. WO 2013169979 A2 20131114.
b MacDonald, R. Loch; Davis, Cara R.; Burton, Kevin; Winchester, Gary; Stella, Angela R. US 20130302431 A1
20131114.。