药物的体内过程
药物的体内过程完整版

药物的体内过程集团标准化办公室:[VV986T-J682P28-JP266L8-68PNN]第三章药物代谢动力学(药动学)药动学(pharmacokinetics)是研究机体对药物的处置过程的科学,即研究药物在体内的吸收、分布、代谢及排泄的过程和血药浓度随时间变化的规律的科学。
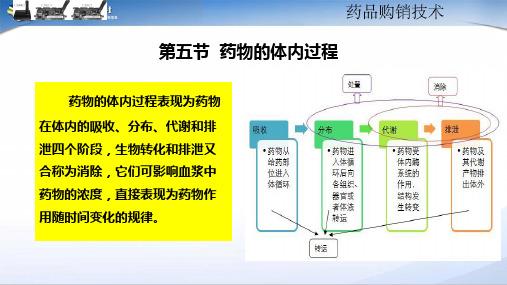
第一节药物体内过程体内过程即吸收(absorption)、分布(distribution)、代谢(metabolism)和排泄(excretion)的过程,又称ADME系统。
吸收、分布、排泄通称药物转运(tranportationofdrug)。
代谢变化也称生物转化(biotransformation)。
代谢和排泄合称为消除(elimination)图3-1药物体内过程示意图一、药物的跨膜转运1.被动转运(passivetransport)类型:1)脂溶扩散(lipiddiffusion;简单扩散)2)水溶扩散(aqueousdiffusion;滤过)3)易化扩散(facilitateddiffusion)(需载体,有饱和、竞争抑制)特点:顺差(浓度、电位),不耗能;不需载体,无饱和、竞争抑制。
2.主动转运(activetransport)特点:逆差(浓度、电位),耗能;需载体,有饱和、竞争抑制。
3.膜动转运(cytopsistransport)胞饮(pinocytosis)胞吐(exocytosis)整个体内过程都涉及药物体内跨膜转运。
大多数药物体内转运过程属于被动转运(脂溶扩散)。
分子量小,非解离型,脂溶性大,极性小的药物易被动转运。
二、吸收药物从给药部位进入血液循环的过程称为吸收。
吸收速度主要影响药物起效的快慢;吸收程度主要影响药物作用的强弱。
影响吸收速度和程度的因素:药物理化性质、剂型、剂量给药途径:起效:吸入>肌内注射>皮下注射>口服>直肠>皮肤吸收环境等。
1.消化道吸收1)口服(oraladministration,peros,p.o.)大多数药物常采用口服给药,以肠道(小肠)吸收为主。
药物在体内的过程PPT课件

药物在细胞内的分布
药物在细胞内的分布主要受到细胞膜通 透性和细胞内药物代谢酶等因素的影响。
细胞膜通透性是指细胞膜对药物的通透 能力,通透性越高,药物进入细胞内的
量越多。
细胞内药物代谢酶是指细胞内对药物进 行代谢的酶类物质,酶的活性越高,药 物在细胞内的代谢越快,从而影响药物
的疗效和安全性。
一些挥发性药物可以通过肺排泄,以气体形式从呼吸道排出体外。 皮肤排泄是指一些药物可以通过皮肤排泄,以汗液或皮肤排泄物的形式排出体外。
药物的肺和皮肤排泄速率取决于药物的理化性质以及排泄途径的通透性。
05
药物效果与副作用
药物的效果
药物的效果是指药物在体内产生的治疗作用, 是药物作用的具体体现。
药物的效果主要取决于药物的性质、用药剂量、 用药方式以及个体差异等因素。
药物的肝脏排泄速率取决于药物的代谢速率和胆汁的 分泌量。
单击此处添加正文,文字是您思想的提一一二三四五 六七八九一二三四五六七八九一二三四五六七八九文 ,单击此处添加正文,文字是您思想的提炼,为了最 终呈现发布的良好效果单击此4*25}
一些药物可能经过肝肠循环而被重复吸收,从而影响 其排泄过程。
药物的肺和皮肤排泄
03
药物代谢
药物的水解
总结词
药物的水解是药物代谢的一种重要过程,涉及到药物的化学结构在酶的作用下 水解成更简单的化合物。
详细描述
药物的水解通常是由水解酶催化完成的,这些酶广泛存在于人体内,如消化道、 肝脏等。水解酶能够识别并作用于药物的化学结构,将其分解成更简单的化合 物。水解反应通常会降低药物的活性,使其失去药效。
药物经粘膜摄入的优点是起效快、生 物利用度高,常用于紧急治疗或局部 治疗,如鼻腔喷雾剂、阴道栓剂等。
药物的体内过程

药物的体内过程(一)吸收定义:药物自给药部位进入血液循环的过程。
静脉注射和静脉滴注直接进入血液,没有吸收过程。
口服给药影响因素:1.药物方面(1)药物的理化性质:脂溶性、解离度、分子量等。
(2)药物剂型:如水剂、注射剂就较油剂、混悬剂、固体剂、缓释制剂、控释制剂.(3)药物制剂:药物崩解度、添加剂、稳定性、F等。
(4)首关消除(首关效应):指口服给药后,部分药物在胃肠道、肠黏膜和肝脏被代谢灭活,使进入体循环的药量减少的现象。
(5)吸收环境(胃肠方面):①蠕动功能;②吸收表面积、血流量、病理状态等。
(6)其他:胃肠内pH,药物在胃肠中相互作用、食物等对药物的影响。
直肠给药经直肠给药仍避免不了首关消除。
吸收不如口服。
唯一优点是防止药物对上消化道的刺激性。
舌下给药由舌下静脉,不经肝脏而直接进入体循环,适合经胃肠道吸收时易被破坏或有明显首过消除的药物。
如四、注射给药特点是吸收迅速、完全。
适用于在胃肠道易被破坏或不易吸收的药物(青霉素G、庆大霉素);也适用于肝中首过消除明显的药物(硝酸甘油)。
硝酸甘油、异丙肾上腺素。
但有些药物注射后因为注射部位发生理化性质改变,导致吸收障碍和注射部位的不适或疼痛,吸收反而比口服的差(地西泮、地高辛、苯妥英钠)。
吸入给药气体和挥发性药物经呼吸道直接由肺泡表面吸收,产生全身作用的给药方式,如吸入麻醉药(乙醚)等。
经皮吸收仅脂溶性极强的有机溶剂和有机磷酸酯类。
皮肤单薄部位(耳后、胸前区、臂内侧等)或有炎症病理改变的皮肤,经皮吸收增加。
药物加入促皮吸收剂如氮酮等制成贴皮剂或软膏,经皮给药后都可达到局部或全身疗效。
(二)分布定义:指吸入血液的药物被转运至组织器官的过程。
药物作用快慢和强弱取决药物分布进入靶器官的速度和浓度,消除的快慢取决药物分布进入代谢和排泄器官(肝脏、肾脏)的速度。
1.与血浆蛋白结合率特点:①差异性。
②暂时失活和暂时贮存血液中.③可逆性。
④饱和性及竞争性。
由于血浆蛋白总量和结合能力有限,加上结合的非特异性,出现两个问题:①当药物结合达到饱和后,继续增加药量,游离型药物浓度增加,出现药物作用或不良反应增强;②同时使用两种以上的药物时,相互竞争与血浆蛋白结合,使其中某些药物游离型增加,出现药物作用或不良反应增强。
第二章 药物的体内过程、药物剂型及处方

肺脏排泄
肺是气体或挥发性药物的排泄器官,如饮酒后呼出带有 酒味的气体。
唾液腺、汗腺、泪腺
某些药物可从唾液中排出,其浓度与血浆浓度相平行。 另外汗腺、泪腺也能排泄某些药物。
胃肠道排泄
经肠道排泄的药物主要是未被吸收的口服药物、随 胆汁排泄到肠道的药物和由肠粘膜主动分泌排入肠 道的药物。
胃内呈强酸性,碱性药物在胃中几乎全部解离,不吸收。 如碱性药物吗啡(pKa=8)中毒时,在胃(pH=1.5-2.5) 中几乎全部解离,重吸收极少,洗胃可清除胃内药物, 如不洗胃清除,则进入碱性肠道再被吸收。
➢ 药→皮肤→体循环
特点:药物须具脂溶性,需透皮辅剂,吸收少。
(临睡前应用硝酸甘油透皮贴片预防夜间心绞痛发作)
➢ 药→眼、鼻→体循环 特点:吸收快速
பைடு நூலகம்
不同给药途径的吸收速率:
静注>腹腔注(吸入)>肌注 >皮下注>舌下>直肠>口服>皮肤
(二) 药物的分布
指血液循环中的药物跨过各种生理屏障转运到 全身各器官、各组织中的过程。
血脑屏障 (Blood-brain barrier, BBB)
由毛细血管壁和 神经胶质细胞形 成的血浆与脑细 胞之间的屏障和 由脉络丛形成的 血浆与脑脊液之 间的屏障。
胎盘屏障(Placental barrier)
胎盘绒毛与子宫血窦 间的屏障,通透性与 一般毛细血管没有明 显区别,屏障功能弱。 多数母体用药都可进 入胎儿。
舌下含服1-2分钟即出现治疗心绞痛的作用。
肠道外给药
➢ 药→血管(静脉、动脉)特点:无吸收过程。
➢ 药→腹腔→体循环
特点:吸收迅速。
➢ 药→肌肉→体循环
特点:吸收较快速,受注射部位血管分布影响
药物的体内过程

(二)器官血流量(blood flow of organs)
再分布(redistribution) 硫喷妥钠(thiopental sodium)首先分布
到血流量大的脑组织发挥全麻作用,随
后由于其脂溶性高又向脂肪组织转移,
病人迅速苏醒。
(三)组织细胞结合
碘 甲状腺 T3、T4
(四)体液pH和药物的解离度(pKa)
significance of Vd ( Vd意义)
1、可以估计药物的分布范围
Vd <5L 药物基本分布于血浆 ≈15L 药物分布于细胞外液
>100L 药物在组织或器官浓集
2、可以计算出期望药物浓度
所需要的给药量。
Vd = A C0
3 )clearance, CL(清除率)
The volume of plasma cleared of drug per unit time is known as the clearance.
的有效成分的生物利用度无显著差
别,则称为生物等效。
ห้องสมุดไป่ตู้
test of bioequivalence
(生物等效性检验)
为了检验药物制剂与参比品在吸收利
用的程度上是否一致,保证药物制剂的安 全可靠性,特地规定药物制剂的AUC、 Tmax及Cmax应在参比品的80~120 %范围 内,称为“等效性检验”。其中AUC等效
特点( characters ):
1)特异性低
(metabolize many drugs) 2)具有个体差异
(have individual variation)
3)具有多种功能(represent a mixedfunction oxidase system) 4)可以被诱导或抑制 (can be induced or inhibited)
药物的体内过程
3.皮肤黏膜和呼吸道的吸收
外用药物时由于皮肤角质层仅可使 脂溶性高的药物通过,皮脂腺的分泌 物覆盖在皮肤表面,可阻止水溶性物 质通过,所以完整皮肤的吸收能力差。 但脂溶性很高的药物可经皮肤吸收, 如硝酸甘油、敌百虫。
呼吸道给药主要由肺泡吸收,肺泡血 流丰富且表面积较大,吸收极其迅速, 凡气体或挥发性药物可直接进入肺泡; 药物溶液经喷雾器雾化后,可到达肺 泡迅速吸收。
化学反应
非氧化还原反应,如酸碱反 应和沉淀反应等;
氧化还原反应,反应过程中 有电子的转移(或电子对的 偏移),反应物和生成物在 前后的化合价发生了改变。
2 Part 氧化还原反应的特点
氧化还原反应的概念: 在反应中有元素化合价变化的化学反应。 (1)实质:有电子的转移(得失或偏移); (2)特征:有元素化合价升降;
2.注射部位的吸收
临床常用的皮下或肌内注射,药液沿结 缔组织或肌纤维扩散,穿过毛细血管壁进 入血液循环。其吸收速度与局部血流量和 药物制剂有关。由于肌肉组织血管丰富、 血流供应充足,故肌内注射较皮下注射吸 收快,休克时周围循环衰竭,皮下或肌内 注射吸收速度减慢,需静脉给药方能即刻 显效。静脉注射时无吸收过程。
2 Part 氧化还原反应的特点
重要的氧化剂一般有以下几类:
活泼的非金属单质,如Cl2、Br2、O2等; 元素处于高化合价时的氧化物,如MnO2等; 元素处于高化合价时的含氧酸,如浓H2SO4 、HNO3等; 元素处于高化合价时的盐,如KMnO4、KClO3 、FeCl3等; 过氧化物,如Na2O2、H2O2等;
2 Part 氧化还原反应的特点
氧化反应(化合价升高) 的实质是原子失去电子的过程。 还原反应(化合价降低) 的实质是原子得到电子的过程。
药理学药物的体内过程

DH为还原药物;DHO为药物代谢产物
该酶的特性:
①专一性低,药物间有竞争性;
②个体差异大;
③酶活性有限;
④其活性一手药物的诱导或抑制
编辑ppt
13
细胞色素P450编在辑药ppt 物氧化中的循环
14
① 药酶诱导剂:能增强P450酶系统活性或增加药
酶生成的药物编辑ppt15药酶抑制剂:能抑制药酶活性或减少要药酶成的
编辑ppt
2
编辑ppt
3
编辑ppt
4
编辑ppt
5
药物的跨膜转运方式
整个体内过程都涉及药物体内跨膜转运 大多数药物体内转运过程属于被动转运(脂溶扩散) 分子量小、非解离型、脂溶性大、极性小的物质易被被动转运
过程一:吸收
吸收(Absorption):药物从给药部位经过细胞屏障膜进入血
液循环的过程称为吸收
物质
编辑ppt
16
过程三:生物转化
非微粒体酶:存在于胃肠道上皮、肾肺血浆。甚至回肠、
结肠的具有转化功能的厌氧菌(统称非微粒体酶)
编辑ppt
17
过程四:排泄
排泄:指药物原型及其代谢物从排泄器官排泄的过程
排泄方式:
一.肾脏排泄: ① 肾小球滤过:主要排泄是小分子物质及未与血浆蛋白结合的药物
② 肾小管再吸收:主要排泄脂溶性高、极性低及非离子型药物
药物的体内过程
2013级生物技术 1324410011 刘静
编辑ppt
1
药物自进入机体至离开可分为以下4个过程 一、吸收-------转运 二、分布-------转运 三、生物代谢转化
四、排泄-------转运 也简称ADME系统即Absorption、 Distribution、Metabolism、Excretion
药物的体内过程

缺点:组织损伤、疼痛、潜在并发症、不
良反应出现迅速,处理相对困难;
适用:需要药物迅速发生作用,因各种原
因不能经口服药的病人。
11
静脉注射:无吸收过程
病情急重多采用
静脉滴注立即可产生药效,并可以控制 用药剂量。
12
肌肉注射和皮下注射: 约30min内达到峰值,吸收速率取决 于注射部位的血管分布状态。
34
肝功能不全时,药酶生成不足或活性降 低,药物转化阻碍。 药酶有种属差异,如家禽无氧化酶,用 巴比妥麻醉时间持久。
35
(四)排泄
是指药物的代谢产物或原形通过各 种途径从体内排出的过程。
36
1、肾排泄:是药物的主要排泄途径
(1)竞争性抑制:青霉素与丙磺舒 ( 2 )利用重吸收原理解毒急救或增强药效,通过 调节尿液 pH 进行。 如 酸性药物在碱性尿液中高度解离、
衡。
22
药物与血浆蛋白结合的特点
可逆性
饱和性
竞争性抑制现象
23
2、组织屏障
血脑屏障—巴比妥类、氯丙嗪类易通过
胎盘屏障—麻醉药、士的宁、拟胆碱药、 抗胆碱药易通过
24
由于药物分布和储存于脂肪、肝、肾、 肌肉和骨骼等组织,这些组织常作为药物残 留的检测材料。
25
(三)生物转化
1、概念:药物在体内经化学变化生成代谢产
二、药物的体内过程
药物的体内过程:吸收、分布、生物转化和排泄 吸收、分布----机体对药物的处置
生物转化和排泄----消除
1
(一)吸收
概念:药物从用药部位进入血液循环的过程。 药物的分子量越小,脂溶性越大或非解离型的比值
越大,越易吸收。 除静脉注射直接进入循环外,药物吸收的快慢、多
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药物的体内过程(ADME)
药物在体内的吸收、分布、代谢和排泄过程首先都要通过细胞膜。
跨膜转运的速度直接影响药物的体内过程,跨膜转运的方式主要有被动转运(passive transport)和主动转运(active transport)两种。
(一)细胞膜的化学成分及结构
细胞膜主要是由按照一定规律排列的脂质、蛋白质及少量的糖类等化学成分构成。
1.膜脂主要有磷脂、糖脂和胆固醇。
2.膜蛋白分为嵌入蛋白(70%—80%)和表在蛋白(20%—30%)两类。
膜蛋白往往充当受体、载体、通道及
酶的作用,在细胞间的识别、物质的跨膜转运及跨膜
信号转导等方面起着重要的作用。
3.膜糖类多为寡糖和多糖链,大都与膜蛋白或膜脂结合形
成糖蛋白或糖脂,分布在质膜外表面,首先与外来刺激相接触,具有受体及抗原的功能。
(二)细胞膜的物质转运功能
1.被动转运是指物质分子或离子顺着浓度梯度或电化学梯度进行的跨膜转运,不需要消耗能量。
(1)简单扩散药物利用生物膜的脂溶性,进行顺浓度差的跨膜转运。
(2)易化扩散借助于膜内特殊载体逆浓度转运。
2.主动运输药物借助细胞膜上的特异性载体,由低浓度侧向高浓度侧的转运过程,需要能量。
一、吸收
概念:药物由给药部位进入血液循环的过程。
口服给药方便,且多数药物能在消化道充分吸收,是常用的给药途径。
根据药物种类不同,可在消化道不同部位吸收,如硝酸甘油可经口腔黏膜吸收,阿司匹林可经胃黏膜吸收,但药物吸收主要在小肠。
小肠的
吸收面积大且肠道内适宜的酸碱
度对药物解离影响小,均有利于
药物在小肠的吸收。
大多数药物在胃肠道内以单
纯扩散方式被吸收。
从胃肠道吸
收入门静脉系统的药物在到达全
身血液循环前先通过肝脏,在肝
脏代谢转化后经血液到达相应的
组织器官发挥作用,最终经肾脏
从尿中排出或经胆汁从粪便排
出。
如果肝脏对药物的代谢能力
强或胆汁排泄量大,会使进入全
身血液循环的有效药量明显减少,因此,凡是在肝脏易凡是
在肝脏易于代谢转化而被
破坏的药物,口服效果差,
以注射为好。
而经舌下及
直肠途径给药,由于药物
不经过门静脉即进入全身
血液循环,避免了药物被肝脏代谢而导致的对药效的影响。
注射部位吸收,临床常采用肌内注射(im)和皮下注射(sc),其吸收速率取决于注射部位的血流量,一般水溶性药物吸收快。
肺部吸收,挥发性或气体性药物通过肺上皮细胞或器官黏膜吸收,可避免首过消除。