常染色体显性遗传病常见的并发症有哪些,怎么根治!
马凡综合症克氏综合征

马凡综合症克氏综合征马凡综合症(Marfan syndrome)是一种罕见的常染色体显性遗传病,也称为克氏综合征(KHS),以其独特的体格特征和多系统受累而著称。
它得名于法国儿科医生安东尼·马凡(Antoine Marfan),他于1896年首次描述了这种病症。
马凡综合症的主要特征是身材高大,肢体延长,骨骼畸形,眼部异常和心血管系统病变。
本文将详细介绍马凡综合症的病因、临床表现、诊断及治疗等方面的知识。
马凡综合症的病因是由于染色体15号上FBN1基因的突变引起的。
该基因编码的蛋白质Fibrillin-1是一种结构性蛋白,主要存在于弹力纤维中,起到维持组织结构和力学性能的重要作用。
FBN1基因突变导致Fibrillin-1蛋白异常,使得弹力纤维的形成和功能发生变化,进而导致马凡综合症的发生。
马凡综合症的临床表现是多样的,主要影响身材、眼部和心血管系统。
患者通常身材高大、肢体细长,手指和脚趾可呈马蹄状。
眼部异常包括近视、角膜薄弱、晶状体脱位等。
心血管系统受累最为显著,患者常伴有心脏杂音、心悸、胸痛等症状,严重者可发展为主动脉瘤或主动脉夹层,后果严重。
诊断马凡综合症的标准主要是根据Ghent 2会议的诊断标准,包括临床表现和家族史,以及特异性的遗传学检测。
根据这些标准,医生可以对马凡综合症进行准确定义的诊断。
目前,马凡综合症的治疗是多学科综合治疗的结果,包括药物治疗、手术治疗和康复治疗等。
药物治疗主要是用于控制症状和预防并发症的发生,如使用β受体阻滞剂来减轻心血管系统的负荷。
手术治疗主要针对心血管系统的疾病,如主动脉瘤或主动脉夹层的修复。
康复治疗则是通过运动和物理疗法来改善患者的肌肉力量和关节灵活性,以提高其生活质量。
虽然马凡综合症是一种遗传性疾病,但是经过及时和有效的治疗,患者的预后可以得到显著改善。
然而,由于马凡综合症的多系统受累性质,患者需要长期的随访和监测,以便及时发现并处理相关的并发症。
马凡氏综合征早期手指

马凡氏综合征早期手指马凡氏综合征(Marfan syndrome)是一种常染色体显性遗传疾病,主要表现为身材高大、四肢异常细长、胸骨凹陷、心脏病变、眼部异常和关节过松。
该综合征早期最常见的体征之一是手指异常,故为手指最早出现症状之一。
本文将详细介绍马凡氏综合征早期手指异常的症状、病因、诊断和治疗。
马凡氏综合征早期手指异常主要表现为手指过长、细长和关节过松。
患者的手指通常比同年龄人更长,并且指骨呈现异常的细长形态。
关节过松则导致患者手指关节松弛,可呈现出虚浮的感觉。
除此之外,患者的手指还容易出现拇指外展畸形,即拇指向外翻。
马凡氏综合征是由于FBN1基因突变引起的。
FBN1基因编码的蛋白质叫做纤维连结蛋白1(fibrillin-1),它是一种构成弹性纤维的关键蛋白质。
FBN1基因突变会导致弹性纤维的合成缺陷,从而影响多个器官的结构与功能。
目前已经发现马凡氏综合征与多个不同的FBN1基因突变相关。
诊断马凡氏综合征需要通过检查患者的临床表现、家族史和遗传学检测。
除了手指异常,马凡氏综合征患者通常还表现出身材高大、四肢细长、胸骨凹陷、驼背、平足等特征。
眼科检查也是确诊马凡氏综合征的重要手段,因为患者的眼部常常出现异常,如近视、晶状体脱位、青光眼等。
治疗马凡氏综合征主要是针对其相关症状进行干预。
对于手指异常,常规的保守治疗措施包括关节支撑物、物理治疗和有针对性的康复训练。
这些方法可以帮助患者改善手指功能并减轻疼痛。
在一些特殊情况下,如严重的手指畸形或功能损害,可能需要进行手术矫正。
除了手指异常,马凡氏综合征在其他器官上还会引起一系列病变。
心脏病变是马凡氏综合征最常见和最严重的并发症之一,包括主动脉扩张和二尖瓣脱垂等。
这些心脏病变可能导致心律失常、主动脉夹层或主动脉瘤的形成,严重者可能导致猝死。
因此,马凡氏综合征患者需要经常进行心脏监测和定期进行心脏影像学检查。
总之,马凡氏综合征是一种常染色体显性遗传疾病,其早期最常见的体征之一是手指异常。
马凡氏综合征诊疗指南

马凡氏综合征诊疗指南马凡氏综合征(Marfan syndrome,MFS)是一种常染色体显性遗传病,主要特征是身材高大、四肢细长、关节松弛、眼底改变及心血管畸形。
马凡氏综合征涉及多个器官系统,其中心血管系统的损害是其主要并且最危险的表现之一。
本文主要内容包括马凡氏综合征的病因、临床特点、诊断标准及治疗指南。
一、病因马凡氏综合征的病因主要是由于FBN1基因突变所致。
FBN1基因编码的是纤维连结蛋白(Fibrillin)-1,该蛋白在胶原纤维中起到支撑和构建作用。
由于FBN1基因突变,导致纤维连结蛋白-1的合成和功能异常,进而影响胶原纤维的形成和结构,导致马凡氏综合征的发生。
二、临床特点马凡氏综合征的临床特点非常多样化,主要表现为以下方面:1. 身材高大、四肢细长:患者身高明显超过同年龄人群,四肢细长而柔软,表现为马凡氏综合征的典型特征。
2. 关节松弛:患者关节过度松弛,易于脱臼,尤其是肩、肘、膝关节。
3. 眼底改变:马凡氏综合征患者眼底可出现玻璃体脱离,视网膜脱离等病变,严重时可导致视力下降。
4. 心血管畸形:马凡氏综合征最危险的并发症是心血管的畸形。
常见的心血管畸形有主动脉扩张、二尖瓣脱垂、主动脉瓣反流等。
这些畸形使得主动脉壁变得薄弱,易于破裂导致主动脉夹层。
主动脉夹层是马凡氏综合征患者的主要死因之一。
5. 其他部位的畸形:马凡氏综合征患者还可表现为下颌突出、齿齿不齐、脊柱侧弯、胸廓畸形等。
三、诊断标准马凡氏综合征的诊断主要是根据临床表现和家族史,同时需要排除其他类似疾病。
由于马凡氏综合征临床表现多样化,诊断标准一般根据国际上统一制定的“Ghent”标准进行评估。
该标准主要包括两个层面:系统的表征和确认的突变。
系统的表征主要包括以下几个方面:身材高大(系统评分2分)、眼底改变(系统评分2分)、心血管畸形(系统评分7分)等。
根据总分,大于等于7分的患者可以确诊为马凡氏综合征。
四、治疗指南马凡氏综合征是一种多系统受累的疾病,因此治疗应综合考虑各个系统的异常表现。
常染色体显性遗传性多囊肾病
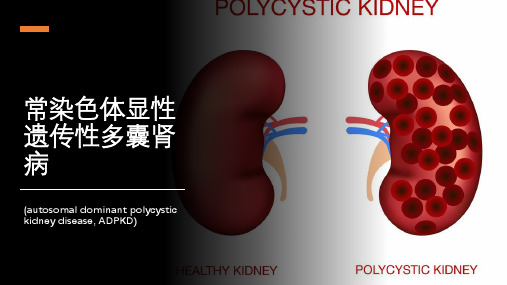
流行病学ห้องสมุดไป่ตู้
•
所有种族都可发生ADPKD,报道的患病率为1:1000。在美国每年
开始透析的患者中,大约有5%的肾病由ADPKD引起。
•
ADPKD主要由两种基因突变引起:16号染色体上的PKD1(编码多
囊蛋白-1)或4号染色体上的PKD2(编码多囊蛋白-2)。大多数患者可维持
正常肾功能到30岁左右。一旦GFR开始下降,其每年可平均降低4.4-
➢ 多囊肝 多数不需治疗。肝脏明显增大可引起腹胀、呼吸困难、胃食管反流、门静脉高压等。可根据病情选 择肝囊肿穿刺硬化、去顶减压术、肝部分切除术或肝移植术
➢ 颅内动脉瘤 对于有动脉瘤和蛛网膜下腔出血家族史的病人,推荐MRI血管造影检查确诊。直径大于10mm的动 脉瘤应采取介入或手术治疗
治疗
肾脏替代治疗 ➢ 包括血液透析、腹膜透析和肾移植。目前认为ADPKD病人腹膜透析与血液透析的并发症和 长期存活率无明显差异。移植后肾存活率、并发症与其他肾移植人群相似
治疗
对症治疗 ➢ 疼痛
急性疼痛针对病因进行治疗。慢性疼痛,程度轻者或一过性疼痛卧床休息并观察,如疼痛持续或较 重按止痛阶梯序贯药物治疗,仍不能缓解可考虑囊肿穿刺硬化、囊肿去顶减压术及多囊肾切除术
➢ 出血 多为囊肿出血所致,呈自限性,轻者绝对卧床休息、止痛、多饮水。出血量大、保守疗法效果差可 行选择性血管栓塞或出血侧肾脏切除
泌尿道和囊肿感染,引起ADPKD病人发热的首要病因,主要表现为膀胱炎、肾盂肾炎、囊肿感 染和肾周脓肿,逆行性感染为主要途径
临床表现
肾外表现 ➢ 囊性病变
可累及肝脏、胰腺、脾脏、卵巢及蛛网膜等器官。其中肝囊肿最常见,大多数病人无症状,少数 可表现为疼痛、囊肿感染和出血
马凡氏综合征有哪些表现

马凡氏综合征有哪些表现马凡氏综合征(Marfan syndrome,MFS)是一种常染色体显性遗传病,主要由于纤维连接蛋白(fibrillin)基因突变引起的结缔组织异常造成的遗传性全身多系统疾病。
该病在全球范围内患病率约为1/5000-1/10000。
马凡氏综合征的主要特征是身材高大、上、下肢异常细长、关节过度活动、视网膜脱离、心血管异常等,严重者可以出现动脉瘤、心脏瓣膜关闭不全、心脏杂音及心律失常等,还伴有一系列的眼、牙等其他系统的病变。
1. 骨骼系统表现:马凡氏综合征的患者身材高大,形象瘦长,比同年龄的正常人高10~20厘米,躯体和四肢发育不平衡。
表现为上、下肢异常细长,手指和脚趾长度超过正常人,手指可超出掌心。
此外,患者还常有驼背、呈非对称性侧弯的脊柱畸形,如脊椎侧弯、躯干和四肢长度不对称等。
2. 心血管系统表现:心血管病变是马凡氏综合征的主要死亡原因。
马凡氏综合征患者常见的心血管问题包括主动脉根部扩张、瓣膜关闭不全以及动脉瘤。
主动脉根部扩张是马凡氏综合征的主要标志之一,会导致主动脉瓣关闭不全。
此外,马凡氏综合征患者还容易出现其他心血管疾病,如二尖瓣脱垂、主动脉瘤、缺血性心脏病、心肌病等。
3. 眼科表现:马凡氏综合征的眼科表现包括近视、视网膜脱离、晶状体脱位等。
患者近视的严重程度在不同个体间有所差异,但一般均较为显著。
视网膜脱离是马凡氏综合征最常见的眼科问题之一,可以引起视力丧失。
晶状体脱位也是马凡氏综合征的特征之一,常导致屈光不正。
4. 呼吸系统表现:马凡氏综合征可引起胸廓异常,导致呼吸功能异常。
患者可出现胸骨凹陷、肋骨变形等。
此外,马凡氏综合征还可以引起气管扁平狭窄、气管食管瘘等。
5. 泌尿生殖系统表现:马凡氏综合征患者的泌尿生殖系统也会受到影响。
男性患者可出现精囊和输精管扩张,女性患者可出现子宫颈松弛。
此外,马凡氏综合征还可以导致肾脏功能异常以及膀胱问题等。
6. 其他系统表现:除了以上主要系统的表现外,马凡氏综合征还可累及其他系统。
马氏凡综合征米津玄师

马氏凡综合征米津玄师马氏凡综合征,又称Marfan综合征,是一种常染色体显性遗传病,以系统性结缔组织异常为主要临床特征。
米津玄师,是日本的一位歌手、词曲创作人和音乐制作人,以其独特的音乐风格和深情的歌曲而受到广泛喜爱。
在接下来的6000字以上篇幅中,我将主要介绍马氏凡综合征以及米津玄师,并探讨其之间的关联。
第一部分:马氏凡综合征的概述1.1 什么是马氏凡综合征?马氏凡综合征是由于一种基因变异导致的组织退行性疾病。
它主要影响身体的结缔组织,包括骨骼、眼睛、心血管系统和皮肤等。
这种遗传病通常是由父母中的一方传递给子女,在一些罕见情况下,也可以是新发生的基因突变。
1.2 马氏凡综合征的症状马氏凡综合征的症状多种多样,下面列举一些常见的症状:- 长而瘦的身材- 高个子- 蜘蛛状手指,也称之为阜万指- 瘦长的四肢- 弓形背- 眼睛问题,如近视、斜视和青光眼等- 心血管问题,如主动脉扩张和瓣膜脱垂等1.3 马氏凡综合征的治疗目前,马氏凡综合征无法治愈,但可以通过治疗来减轻症状和管理并发症。
常见的治疗方式包括:- 药物治疗:用药物控制心血管问题、眼睛问题和其他症状。
- 手术干预:对于严重的心血管问题和眼睛问题,可能需要进行手术。
- 生活方式管理:包括定期进行体检、保持健康饮食和适度的锻炼等。
第二部分:米津玄师的简介2.1 米津玄师的背景米津玄师,本名低音质修太(Kentaro Daisuke),于1991年出生于日本兵库县。
他是日本知名音乐厂牌TOY'S FACTORY 的旗下签约艺人,也是日本国内非常受欢迎的流行音乐创作人之一。
2.2 米津玄师的音乐风格米津玄师的音乐风格非常独特,将流行、摇滚、电子和民谣等元素融合在一起,形成了独特的声音,并在日本乐坛上树立了自己的地位。
他的歌曲引人入胜,旋律优美,歌词深情,常常带给听众强烈的情感共鸣。
2.3 米津玄师与马氏凡综合征的关联据一些报道,米津玄师在早年被怀疑患有马氏凡综合征,因为他具有身材高大瘦长的特点,以及指关节异常。
我是大医生马凡氏综合征

我是大医生马凡氏综合征我是大医生,今天给大家介绍一种罕见的遗传疾病——马凡氏综合征。
马凡氏综合征(Marfan syndrome)是一种遗传性结缔组织疾病,以身材高大、骨骼发育异常、心血管系统病变为特征。
这个病名来源于其首位发现者法国儿科医生安东尼·马凡。
马凡氏综合征是一种常染色体显性遗传病,该病的发病率约为每3,000到1万人中有一人。
马凡氏综合征的临床表现多种多样,主要有身材高大、肢体比例异常、眼部病变和心血管系统异常等。
患者身高通常超过同龄人,手指和趾趾非常细长,伸出手时指关节呈现过度屈曲的形态,脸部特征也有明显的变化,例如下颌明显后缩,上颌过度突出等。
马凡氏综合征的眼部表现主要包括近视、角膜脱垂、晶状体脱位等。
角膜脱垂是马凡氏综合征的一个重要特征,患者常常出现眼压增高或者青光眼,严重者还可能发生视网膜脱离。
而晶状体脱位是马凡氏综合征中较常见的眼部病变,导致患者视力严重下降。
心血管异常是马凡氏综合征中最危险的并发症之一。
患者常常出现主动脉瓣关闭不全、动脉瘤形成等病症。
主动脉瓣关闭不全会导致心血液回流,使心脏负荷过重,严重影响患者的生活质量。
而动脉瘤形成则可能引发主动脉夹层,这是一种十分严重的疾病,如果不及时治疗,极易导致患者大出血甚至死亡。
在马凡氏综合征的治疗中,手术是目前最常用的方法。
对于世界上最高的人记录保持者——张腾飞,马凡氏综合征再现的病状是近几年出现的,也是目前唯一一个出现这个病例的中国人。
经过多次手术,张腾飞终于使身高稳定在2米9左右,相对于他6岁的时候长的仅仅1米4的身高来说,可以说是缩短了不少。
传统手术方法主要是通过外科手术修复主动脉夹层、硬化性动脉瘤等病变,以及替换病变的主动脉瓣等。
然而,传统手术方法并不能完全解决马凡氏综合征的问题,需要长时间的恢复期,手术风险也较高。
近年来,随着医学技术的不断发展,心脏介入治疗成为了马凡氏综合征的新治疗方式。
尤其是经导管主动脉内膜修复术(TEVAR),其在改善心功能、缓解主动脉破裂风险等方面表现出了明显的效果。
马凡氏综合征遗传性

马凡氏综合征遗传性马凡氏综合征(Marfan syndrome,MFS)是一种遗传性结缔组织疾病,主要特征是身材高大、四肢细长、关节松弛、眼球脱位以及心血管问题,属于常染色体显性遗传疾病。
其命名来源于法国儿科医生安东尼·柏格(Antoine Marfan),于1896年首次描述了该病的病征。
本文将系统介绍马凡氏综合征的遗传机制、病因、临床表现以及诊断和治疗方法。
一、遗传机制马凡氏综合征是由于FBN1(fibrillin-1)基因突变引起的,该基因位于15号染色体长臂上。
FBN1基因编码的蛋白质是糖蛋白,主要存在于结缔组织中,包括皮肤、韧带、血管等。
该蛋白在细胞外基质形成、维护和修复过程中起着重要作用。
FBN1基因的突变会导致糖蛋白的含量、结构和功能发生改变,进而影响结缔组织的稳定性、弹性和可塑性。
马凡氏综合征的遗传方式为常染色体显性遗传,即一个患者的父母中至少有一人是患病者或是基因突变的携带者。
患有马凡氏综合征的父母有50%的概率将疾病基因传给子代,而如果一位父母是基因突变的携带者,子代有25%的概率患病,50%的概率为携带者,25%的概率正常。
二、病因马凡氏综合征的病因主要是FBN1基因的突变,突变类型包括错义突变、无义突变、插入缺失等。
这些突变会导致FBN1基因表达有缺陷,糖蛋白的合成和分泌减少,进而影响结缔组织的形成和功能。
此外,近年来的研究还发现,除FBN1基因突变外,其他与马凡氏综合征相关的基因也可能参与疾病的发生和发展,包括TGFBR2、TGFBR1、COL3A1等。
对于马凡氏综合征发病机制的理解,科学家提出了两种主要的假说,即结构假说和生物信号假说。
结构假说认为,FBN1蛋白的缺陷会导致结缔组织中的纤维网状结构松弛和脱离,进而引发马凡氏综合征的临床表现。
生物信号假说认为,FBN1蛋白对TGF-β信号通路起调节作用,在缺陷的情况下,TGF-β信号通路失控,影响细胞增殖、增生和分化,从而导致结构改变和组织异常。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
常染色体显性遗传病常见的并发症有哪些,怎么根治!常染色体显性遗传病常见的并发症常染色体显性遗传病有什么并发症一、并发病症常染色体显性遗传病主要包括以下几种病征。
1、软骨发育不全其主要特征为四肢短小畸形,可能系遗传性侏儒症中最常见的类型。
因长骨骺端软骨细胞形成障碍,影响骨的长度,但骨的宽度仍然增长,而导致四肢短小,侏儒体型。
出生时即呈现四肢短而粗,躯干相对较长;手指短而粗,各指长度相仿,两手下垂不过髋关节;儿童期或成年后头部明显过大,前额突出,马鞍鼻,颏部大而前突。
此外,尚有腰椎前凸或驼背,两下肢内弯,步态摇摆,X线检查长骨变短,弯曲,两端膨大,头颅和前盆均具特征。
患者智力及生殖功能正常。
女性患者妊娠后,因骨盆狭窄需剖宫分娩。
本症为常染色体显性遗传,故子代中有半数发病机会,但有相当一部分患者为基因突变所致。
有人认为此基因突变与父龄过高有关。
若患者为纯合子,则有双倍的基因效应,常可致死。
超声和X线检查可做产前诊断。
微信:39健康百科(微信号:jibingbaike39),可了解该疾病更多治疗方法,发病原因,护理知识。
二、蜘蛛脚样指综合征其特征为肢体过长,眼病和心血管异常。
因长骨过度生长而呈身材细长体型,上下段比例失常,四肢长,尤其指、趾细长;肋骨异常呈漏斗胸;肌肉发育差,皮下脂肪少,关节松弛;晶状体异位或重度近视;主动脉瓣关闭不全或主动脉瘤。
主动脉瘤破裂是早亡的主要原因,妊娠增加主动脉破裂的危险,尤其在分娩前后,虽无主动脉瘤,但妊娠后亦应作为高危妊娠监护。
本征虽为常染色体显性遗传,但有时轻症者表现不典型而又无其他重症亲属可见时,则轻度患者无法确诊,咨询中发生困难。
3、多发性神经纤维瘤病本病为神经外胚层的病变,皮肤上有黄棕色的色素斑为典型的特殊体征,呈卵圆形或环状不一,直径为1~5cm;还常伴有皮肤神经纤维瘤,呈多发性,较小,质柔软,稀疏分布,大的神经纤维瘤常在外周神经或神经根上,可导致脊柱畸形。
中枢神经系统的神经纤维瘤最常累及听神经,若有弥漫性病变则伴有轻度智力障碍。
本病有伴发恶性肿瘤的危险,神经纤维瘤或其他部位均可并发恶性肿瘤,其几率为10%~20%。
尚有脊柱侧凸、轻度智力障碍、癫痫、神经根压迫、胫骨假关节和嗜铬细胞瘤等。
三、遗传性舞蹈病本病为大脑中基底核的病变,主要表现为进行性痴呆和不自主的舞蹈动作。
起病隐匿,仅是正常的面部动作和手势增加,以后呈现舞蹈样的不随意运动,舞蹈样动作缓慢,两次动作的间歇期较长。
早期诊断较为困难,虽能发现基底核的萎缩变化,但达不到早期诊断的要求。
本病发病年龄在40 岁左右,少数可在儿童后期发病,早期即有智力减退,呈进行性痴呆,大多于10年后恶化。
本病虽为常染色体显性遗传,但遗传咨询常达不到预期效果,因为患者大多于婚配且生育后发病,而其子女中虽有一半机会再患此病,但无法预测亦无法早期诊断,一般要到40岁尚未发病,方能认为基本上摆脱了发病危险。
5、地中海贫血本病为血红蛋白的一种或多种肽链合成量减少而引起的遗传性血红蛋白病。
按Bart's水肿胎儿综合征。
因二者有不同的组合可产生不同轻重的病情,以HbBart's水肿胎儿综合征最为严重,而孕妇常有水肿和高血压。
其他病情较轻者常出现贫血及其引起的心肺和造血功能异常的表现。
轻症者可无症状,不必治疗。
②Bart's水肿胎儿的产前诊断。
6、家族性高脂蛋白血症(hypercholesterolaemia)高脂蛋白血症有原发性和继发性两类,原发性患者多为遗传的。
高脂蛋白血症在生物化学上可分5型,其中Ⅱ型及Ⅲ型与冠状动脉粥样硬化性心脏病关系最密切。
高脂蛋白血症Ⅱ型患者的生化特点是血浆中粥样硬化。
此后有纤维组织增生,并可有钙质沉着,或发生局部血栓形成,内膜凹凸不平,管腔狭窄,使管壁硬化。
若硬化发生于大动脉,不会影响血液供应,若发生于中型动脉(如冠状动脉、脑动脉、肾动脉等),则引起相应脏器供血不足,甚至发生梗阻。
所以高脂蛋白血症Ⅱ型的最危险并发症是早发的冠状动脉粥样硬化,常并发心绞痛与心肌梗塞。
此病常并发黄色瘤,尤其常见的是睑黄斑瘤,致病基因定位于19p13.2~p13.1。
五、其他(Wilsons syndrome)致病基因携带者在10岁之前一切发育正常,往往在10~20岁之间突然发作,出现脑中心退化,肝细胞被纤维组织代替造成肝硬化,角膜中间出现色环,眼球震颤,肌张力亢进,尿中含大量末端双羧氨基酸的肽和氨基酸残基等。
它是由于基因突变引起患者体内铜代谢障碍所致。
9、亨丁顿氏舞蹈病(huntingtons disease)此病是一种完全符合孟德尔氏遗传的显性遗传病。
患者20岁前很少发病,20岁后发病率逐渐增高。
发病时,最初表现为情绪波动,随后出现舞蹈性动作,癫痫发作,体力和智力不断减退,进行性痴呆。
常于症状出现后的4~20年间死亡。
此病有明显的家族遗传史,只要双亲之一是患者,他们的子女中至少会有1/2的发病机率。
10、结肠息肉(peutz jeghers syndromeⅠ)此病有明显家族遗传倾向,患者最早可在20岁左右发生恶变,结肠上长有大小不等的肉瘤,引起胃肠出血和腹泻,息肉恶变的可能性较大,需进行结肠切除手术。
同此病相类似的另一种肠道遗传病是空肠息肉(syndromeⅡ),患者的早期在口、唇周围及口腔粘膜和手指上面出现色素斑点,到成年则有消退的趋势。
良性息肉主要分布在空肠,但也偶发在肠道的其他部位,或膀胱及呼吸道,伴有腹部绞痛、胃肠出血和肠套叠等并发症。
儿童期可发病。
11、阵发性心动过速(paroxysmal tachycardia)此病有明显的家族史,可连续几代遗传。
能在任何年龄阶段出现阵发性心动过速,病理多属器质性的,也有纯功能性的。
12、体质性低血压此病患者常见于体质瘦弱者,女子多于男子。
多数患者无自觉症状,少数有疲倦、健忘、头晕、头痛等。
这些症状常因合并某些疾病或营养不良所致。
13、椭圆形红细胞增多症(hereditary elliptocytosis)此病是有两个不同位点上的基因各自控制的疾病,患者有50%或更多的红细胞呈椭圆形、卵圆形、香肠形和杆状(正常人最多为10%),这种异形红细胞最早可出现在3~4个月龄婴儿的外周血液循环中。
此病患者在儿童少年多无症状表现,但存在不同程度的溶血,其中包括伴发再生障碍危象的严重溶血形式,而且病人有脾大现象。
14、肌强直性营养不良(steinert disease)此病的遗传表现为母亲如果有病,其子女患病的可能性更大。
有的患者在儿童期发病,而更多的是在成年初期发病。
最常见的症状是颌部和手部肌肉松弛、收缩困难,肌肉萎缩、无力,面容无表情,可发生白内障。
男性有额秃,睾丸萎缩;女性则有闭经,痛经和卵巢囊肿。
患者可有心律不齐、传导缺陷和充血性心力衰竭,也有智力障碍。
15、先天性肌强直(thomsem disease)此病主要症状为普遍性肌强直和肌肥大,多数在出生时或儿童早期即发病,少数至青春期发病。
患者肢体僵硬,动作笨拙,静止不动后或在寒冷环境中症状加重,反复运动可暂时减轻症状。
坐或站立一段时间后,不能立即起立或起步。
突然受惊吓时,可引起全身肌肉的强直性收缩;跌倒时不能将手伸出撑住地面及时爬起;与人握手后要较长时间才能松开。
打喷嚏后,双眼仍紧闭;发笑后,面部表情肌不能及时恢复;温暖的环境能使肌强直减轻。
症状严重程度可因人而异,最轻者甚至无自觉主诉,仅在家系调查中发现。
个别病人在肌肉多次收缩后症状不见减轻,反而加重,称为反常性肌强直。
病人全身肌肉发育良好,常伴肥大。
此病预后良好,多数随年龄增长而症状减轻,对寿命无影响。
16、周期性麻痹(periodic paralysis)此病根据致病基因携带者受刺激后机体的血钾水平表现不同分为不同种类型。
一种类型是在激烈运动后长时间休息、食用高碳水化合物、焦急忧虑、遇到寒冷或服用多种药物(包括胰岛素、肾上腺素、乙醇、一些无机物、皮质激素和甘草属植物)等情况下,可促发机体低血钾麻痹。
而另一种类型是激烈运动时、服用氯化钾以及使用某些麻醉剂可引起高血钾性麻痹。
17、胱氨酸尿症(cystinuria)此病主要原因是肾小管对胱氨酸、赖氨酸、精氨酸、鸟氨酸的重吸收发生障碍所致。
患者尿中有上述4种氨基酸排出,但无任何症状。
由于胱氨酸易生成六角形结晶,故可发生尿路结石(胱氨酸结石)。
尿路结石可引起尿路感染和绞痛。
纯合子患者4种氨基酸排泄量均增加,杂合子患者胱氨酸和赖氨酸排泄量有少量增加。
18、遗传性球形细胞增多症(herediatry spherocytosis)此病是一种慢性溶血性贫血,主要特征是黄疸、脾大、红细胞球形改变、脆性增加。
新生儿期发病时可有严重贫血和黄疸症状,婴儿期除轻度或中度贫血外常无其他症状,幼儿及年长儿发病时,其主要症状是轻度黄疸及贫血。
如过度劳累、受冷感冒可使黄疸加重,伴有发热、呕吐、腹痛、肝脾压痛、无力、心跳加快、气促,脾脏增大可达肋下 2.8cm,肝脏略有增大。
血液学检查可见红细胞直径变小,胞体变圆,网织红细胞增高5%~20%,红细胞脆性增高可达0.40%~0.68%,自身溶血试验呈阳性。
常染色体显性遗传病的种类很多,除上述以外,比较常见的还有软骨发育不全症、短指畸形、肾性糖尿病、先天性白内障、夜盲症、青光眼、视网膜母细胞瘤、先天性眼睑下垂、多指畸形、多囊肾、遗传性神经性耳聋、过敏性鼻炎、牙齿肥大症、多胎妊娠及尿崩症等。
文章来自:39疾病百科 /crstxxycb/bfbz/。